Plink#
In this notebook we will use PLink to caluclate the PRS.
Note: Plink needs to be installed or placed in the same directory as this notebook.
We recommend using Linux. In cases where Windows is required due to package installation issues on Linux, we provide the following guidance:
For Windows, use
plink.For Linux, use
./plink.
Plink Hyperparameters#
Plink is a tool that allows us to perform clumping and pruning. It also lets us specify the p-value thresholds used on the training data. For each combination of clumping, pruning, and p-value thresholds, a polygenic risk code is generated for each person. Plink takes beta coefficients or OR ratios from the GWAS file without re-estimating those values. Clumping and pruning are performed on the training data using the specified p-value thresholds. The same remaining number of SNPs from the test set is then used to estimate the polygenic risk scores. No separate clumping and pruning are required on the test set.
Details about clumping can be found here, and information about pruning is available here. P-value threshold documentation can be found [here](https://www.cog-genomics.org/plink/2.0/score#:~:text=–q-score-range can,in the third column%2C e.g., for the scorecard.
Pruning Parameters#
Informs Plink that we wish to perform pruning with a window size of 200 variants, sliding across the genome with a step size of 50 variants at a time, and filter out any SNPs with LD ( r^2 ) higher than 0.25.
1. p_window_size = [200]
2. p_slide_size = [50]
3. p_LD_threshold = [0.25]
Clumping Parameters#
The P-value threshold for an SNP to be included. 1 means to include all SNPs for clumping. SNPs having ( r^2 ) higher than 0.1 with the index SNPs will be removed. SNPs within 200k of the index SNP are considered for clumping.
1. clump_p1 = [1]
2. clump_r2 = [0.1]
3. clump_kb = [200]
Score Parameters#
–q-score-range can be used to apply –score too many variants subsets at once, based on, e.g., p-value ranges.
The “range file” should have range labels in the first column, p-value lower bounds in the second column, and upper bounds in the third column, e.g.
1. pv_1 0.00 0.01
2. pv_2 0.00 0.20
PCA#
Pca also affects the results evident from the initial analysis; however, including more PCA overfits the model.
Kindly note that the number of p-values to be considered varies, and the actual p-value also depends on the dataset. Moreover, after clumping, pruning, and p-value threshold, the number of SNPs in each fold can vary.
#!pip install statsmodels
#!pip install scikit-learn
GWAS file processing for Plink for Binary Phenotypes.#
When the effect size relates to disease risk and is thus given as an odds ratio (OR) rather than BETA (for continuous traits), the PRS is computed as a product of ORs. To simplify this calculation, take the natural logarithm of the OR so that the PRS can be computed using summation instead.
For continuous phenotype GWAS, the SampleData1/SampleData1.gz file should have BETAs, and for binary phenotypes, it should have OR instead of BETAs. If BETAs are not available, we convert OR to BETAs using BETA = np.log(OR) and convert BETAs to OR using OR = np.exp(BETA).
import numpy as np; np is the NumPy module.
import os
import pandas as pd
import numpy as np
import sys
#filedirec = sys.argv[1]
filedirec = "SampleData1"
#filedirec = "asthma_19"
#filedirec = "migraine_0"
# Check the status of the Phenotype.
# If Phenotype is Binary, Plink requires OR, and if it continous, it requires BETAS in the GWAS file.
def check_phenotype_is_binary_or_continous(filedirec):
# Read the processed quality controlled file for a phenotype
df = pd.read_csv(filedirec+os.sep+filedirec+'_QC.fam',sep="\s+",header=None)
column_values = df[5].unique()
if len(set(column_values)) == 2:
return "Binary"
else:
return "Continous"
# Read the GWAS file.
GWAS = filedirec + os.sep + filedirec+".gz"
df = pd.read_csv(GWAS,compression= "gzip",sep="\s+")
if "BETA" in df.columns.to_list():
# For Continous Phenotype.
df = df[['CHR', 'BP', 'SNP', 'A1', 'A2', 'N', 'SE', 'P', 'BETA', 'INFO', 'MAF']]
else:
df["BETA"] = np.log(df["OR"])
df = df[['CHR', 'BP', 'SNP', 'A1', 'A2', 'N', 'SE', 'P', 'BETA', 'INFO', 'MAF']]
df.to_csv(filedirec + os.sep +filedirec+".txt",sep="\t",index=False)
df.to_csv(filedirec + os.sep +filedirec+"_Plink.txt",sep="\t",index=False)
print(df.head().to_markdown())
print("Length of DataFrame!",len(df))
| | CHR | BP | SNP | A1 | A2 | N | SE | P | BETA | INFO | MAF |
|---:|------:|-------:|:-----------|:-----|:-----|-------:|-----------:|---------:|------------:|---------:|---------:|
| 0 | 1 | 756604 | rs3131962 | A | G | 388028 | 0.00301666 | 0.483171 | -0.00211532 | 0.890558 | 0.36939 |
| 1 | 1 | 768448 | rs12562034 | A | G | 388028 | 0.00329472 | 0.834808 | 0.00068708 | 0.895894 | 0.336846 |
| 2 | 1 | 779322 | rs4040617 | G | A | 388028 | 0.00303344 | 0.42897 | -0.00239932 | 0.897508 | 0.377368 |
| 3 | 1 | 801536 | rs79373928 | G | T | 388028 | 0.00841324 | 0.808999 | 0.00203363 | 0.908963 | 0.483212 |
| 4 | 1 | 808631 | rs11240779 | G | A | 388028 | 0.00242821 | 0.590265 | 0.00130747 | 0.893213 | 0.45041 |
Length of DataFrame! 499617
Define Hyperparameters#
Define hyperparameters to be optimized and set initial values.
Extract Valid SNPs from Clumped File#
For Windows, download gwak, and for Linux, the awk command is sufficient. For Windows, GWAK is required. You can download it from here. Get it and place it in the same directory.
Execution Path#
At this stage, we have the genotype training data newtrainfilename = "train_data.QC" and genotype test data newtestfilename = "test_data.QC".
We modified the following variables:
filedirec = "SampleData1"orfiledirec = sys.argv[1]foldnumber = "0"orfoldnumber = sys.argv[2]for HPC.
Only these two variables can be modified to execute the code for specific data and specific folds. Though the code can be executed separately for each fold on HPC and separately for each dataset, it is recommended to execute it for multiple diseases and one fold at a time. Here’s the corrected text in Markdown format:
P-values#
PRS calculation relies on P-values. SNPs with low P-values, indicating a high degree of association with a specific trait, are considered for calculation.
You can modify the code below to consider a specific set of P-values and save the file in the same format.
We considered the following parameters:
Minimum P-value:
1e-10Maximum P-value:
1.0Minimum exponent:
10(Minimum P-value in exponent)Number of intervals:
100(Number of intervals to be considered)
The code generates an array of logarithmically spaced P-values:
import numpy as np
import os
minimumpvalue = 10 # Minimum exponent for P-values
numberofintervals = 100 # Number of intervals to be considered
allpvalues = np.logspace(-minimumpvalue, 0, numberofintervals, endpoint=True) # Generating an array of logarithmically spaced P-values
print("Minimum P-value:", allpvalues[0])
print("Maximum P-value:", allpvalues[-1])
count = 1
with open(os.path.join(folddirec, 'range_list'), 'w') as file:
for value in allpvalues:
file.write(f'pv_{value} 0 {value}\n') # Writing range information to the 'range_list' file
count += 1
pvaluefile = os.path.join(folddirec, 'range_list')
In this code:
minimumpvaluedefines the minimum exponent for P-values.numberofintervalsspecifies how many intervals to consider.allpvaluesgenerates an array of P-values spaced logarithmically.The script writes these P-values to a file named
range_listin the specified directory.
from operator import index
import pandas as pd
import numpy as np
import os
import subprocess
import sys
import pandas as pd
import statsmodels.api as sm
import pandas as pd
from sklearn.metrics import roc_auc_score, confusion_matrix
from statsmodels.stats.contingency_tables import mcnemar
def create_directory(directory):
"""Function to create a directory if it doesn't exist."""
if not os.path.exists(directory): # Checking if the directory doesn't exist
os.makedirs(directory) # Creating the directory if it doesn't exist
return directory # Returning the created or existing directory
#foldnumber = sys.argv[2]
foldnumber = "0" # Setting 'foldnumber' to "0"
folddirec = filedirec + os.sep + "Fold_" + foldnumber # Creating a directory path for the specific fold
trainfilename = "train_data" # Setting the name of the training data file
newtrainfilename = "train_data.QC" # Setting the name of the new training data file
testfilename = "test_data" # Setting the name of the test data file
newtestfilename = "test_data.QC" # Setting the name of the new test data file
# Number of PCA to be included as a covariate.
numberofpca = ["6"] # Setting the number of PCA components to be included
# Clumping parameters.
clump_p1 = [1] # List containing clump parameter 'p1'
clump_r2 = [0.1] # List containing clump parameter 'r2'
clump_kb = [200] # List containing clump parameter 'kb'
# Pruning parameters.
p_window_size = [200] # List containing pruning parameter 'window_size'
p_slide_size = [50] # List containing pruning parameter 'slide_size'
p_LD_threshold = [0.25] # List containing pruning parameter 'LD_threshold'
# Kindly note that the number of p-values to be considered varies, and the actual p-value depends on the dataset as well.
# We will specify the range list here.
minimumpvalue = 10 # Minimum p-value in exponent
numberofintervals = 20 # Number of intervals to be considered
allpvalues = np.logspace(-minimumpvalue, 0, numberofintervals, endpoint=True) # Generating an array of logarithmically spaced p-values
print("Minimum P-value",allpvalues[0])
print("Maximum P-value",allpvalues[-1])
print("Number of P-value",len(allpvalues))
count = 1
with open(folddirec + os.sep + 'range_list', 'w') as file:
for value in allpvalues:
file.write(f'pv_{value} 0 {value}\n') # Writing range information to the 'range_list' file
count = count + 1
pvaluefile = folddirec + os.sep + 'range_list'
# Initializing an empty DataFrame with specified column names
prs_result = pd.DataFrame(columns=["clump_p1", "clump_r2", "clump_kb", "p_window_size", "p_slide_size", "p_LD_threshold",
"pvalue", "numberofpca","numberofvariants","Train_pure_prs", "Train_null_model", "Train_best_model",
"Test_pure_prs", "Test_null_model", "Test_best_model"])
Minimum P-value 9.999999999999999e-11
Maximum P-value 1.0
Number of P-value 20
Define Helper Functions#
Perform Clumping and Pruning
Calculate PCA Using Plink
Fit Binary Phenotype and Save Results
Fit Continuous Phenotype and Save Results
import os
import subprocess
import pandas as pd
import statsmodels.api as sm
from sklearn.metrics import explained_variance_score
# The following function is used to perform clumping and pruing on the Genotype Data.
# It is almost the same in for all the phenotypes.
def perform_clumping_and_pruning_on_individual_data(traindirec, newtrainfilename,numberofpca, p1_val, p2_val, p3_val, c1_val, c2_val, c3_val,Name,pvaluefile):
command = [
"./plink",
"--bfile", traindirec+os.sep+newtrainfilename,
"--indep-pairwise", p1_val, p2_val, p3_val,
"--out", traindirec+os.sep+trainfilename
]
subprocess.run(command)
# First perform pruning and then clumping and the pruning.
command = [
"./plink",
"--bfile", traindirec+os.sep+newtrainfilename,
"--clump-p1", c1_val,
"--extract", traindirec+os.sep+trainfilename+".prune.in",
"--clump-r2", c2_val,
"--clump-kb", c3_val,
"--clump", filedirec+os.sep+filedirec+".txt",
"--clump-snp-field", "SNP",
"--clump-field", "P",
"--out", traindirec+os.sep+trainfilename
]
subprocess.run(command)
# Extract the valid SNPs from th clumped file.
# For windows download gwak for linux awk commmand is sufficient.
### For windows require GWAK.
### https://sourceforge.net/projects/gnuwin32/
##3 Get it and place it in the same direc.
#os.system("gawk "+"\""+"NR!=1{print $3}"+"\" "+ traindirec+os.sep+trainfilename+".clumped > "+traindirec+os.sep+trainfilename+".valid.snp")
#print("gawk "+"\""+"NR!=1{print $3}"+"\" "+ traindirec+os.sep+trainfilename+".clumped > "+traindirec+os.sep+trainfilename+".valid.snp")
#Linux:
command = f"awk 'NR!=1{{print $3}}' {traindirec}{os.sep}{trainfilename}.clumped > {traindirec}{os.sep}{trainfilename}.valid.snp"
os.system(command)
command = [
"./plink",
"--make-bed",
"--bfile", traindirec+os.sep+newtrainfilename,
"--indep-pairwise", p1_val, p2_val, p3_val,
"--extract", traindirec+os.sep+trainfilename+".valid.snp",
"--out", traindirec+os.sep+newtrainfilename+".clumped.pruned"
]
subprocess.run(command)
command = [
"./plink",
"--make-bed",
"--bfile", traindirec+os.sep+testfilename,
"--indep-pairwise", p1_val, p2_val, p3_val,
"--extract", traindirec+os.sep+trainfilename+".valid.snp",
"--out", traindirec+os.sep+testfilename+".clumped.pruned"
]
subprocess.run(command)
# This function calculats PCA for test and train genotype data, which is then Appended with covariates, PRS, for prediction.
def calculate_pca_for_traindata_testdata_for_clumped_pruned_snps(traindirec, newtrainfilename,p):
# Calculate the PRS for the test data using the same set of SNPs and also calculate the PCA.
# Also extract the PCA at this point.
# PCA are calculated afer clumping and pruining.
command = [
"./plink",
"--bfile", folddirec+os.sep+testfilename+".clumped.pruned",
# Select the final variants after clumping and pruning.
"--extract", traindirec+os.sep+trainfilename+".valid.snp",
"--pca", p,
"--out", folddirec+os.sep+testfilename
]
subprocess.run(command)
command = [
"./plink",
"--bfile", traindirec+os.sep+newtrainfilename+".clumped.pruned",
# Select the final variants after clumping and pruning.
"--extract", traindirec+os.sep+trainfilename+".valid.snp",
"--pca", p,
"--out", traindirec+os.sep+trainfilename
]
subprocess.run(command)
# This function used for prediction of binary phenotypes.
def fit_binary_phenotype_on_PRS(traindirec, newtrainfilename,p, p1_val, p2_val, p3_val, c1_val, c2_val, c3_val,Name,pvaluefile):
threshold_values = allpvalues
# Merge the covariates, pca and phenotypes.
tempphenotype_train = pd.read_table(traindirec+os.sep+newtrainfilename+".clumped.pruned"+".fam", sep="\s+",header=None)
phenotype_train = pd.DataFrame()
phenotype_train["Phenotype"] = tempphenotype_train[5].values
pcs_train = pd.read_table(traindirec+os.sep+trainfilename+".eigenvec", sep="\s+",header=None, names=["FID", "IID"] + [f"PC{str(i)}" for i in range(1, int(p)+1)])
covariate_train = pd.read_table(traindirec+os.sep+trainfilename+".cov",sep="\s+")
covariate_train.fillna(0, inplace=True)
covariate_train = covariate_train[covariate_train["FID"].isin(pcs_train["FID"].values) & covariate_train["IID"].isin(pcs_train["IID"].values)]
covariate_train['FID'] = covariate_train['FID'].astype(str)
pcs_train['FID'] = pcs_train['FID'].astype(str)
covariate_train['IID'] = covariate_train['IID'].astype(str)
pcs_train['IID'] = pcs_train['IID'].astype(str)
covandpcs_train = pd.merge(covariate_train, pcs_train, on=["FID","IID"])
covandpcs_train.fillna(0, inplace=True)
## Scale the covariates!
from sklearn.preprocessing import MinMaxScaler
from sklearn.metrics import explained_variance_score
scaler = MinMaxScaler()
normalized_values_train = scaler.fit_transform(covandpcs_train.iloc[:, 2:])
#covandpcs_train.iloc[:, 2:] = normalized_values_test
tempphenotype_test = pd.read_table(traindirec+os.sep+testfilename+".clumped.pruned"+".fam", sep="\s+",header=None)
phenotype_test= pd.DataFrame()
phenotype_test["Phenotype"] = tempphenotype_test[5].values
pcs_test = pd.read_table(traindirec+os.sep+testfilename+".eigenvec", sep="\s+",header=None, names=["FID", "IID"] + [f"PC{str(i)}" for i in range(1, int(p)+1)])
covariate_test = pd.read_table(traindirec+os.sep+testfilename+".cov",sep="\s+")
covariate_test.fillna(0, inplace=True)
covariate_test = covariate_test[covariate_test["FID"].isin(pcs_test["FID"].values) & covariate_test["IID"].isin(pcs_test["IID"].values)]
covariate_test['FID'] = covariate_test['FID'].astype(str)
pcs_test['FID'] = pcs_test['FID'].astype(str)
covariate_test['IID'] = covariate_test['IID'].astype(str)
pcs_test['IID'] = pcs_test['IID'].astype(str)
covandpcs_test = pd.merge(covariate_test, pcs_test, on=["FID","IID"])
covandpcs_test.fillna(0, inplace=True)
normalized_values_test = scaler.transform(covandpcs_test.iloc[:, 2:])
#covandpcs_test.iloc[:, 2:] = normalized_values_test
# One can use multiple ranges of alphas and weights for Binary Phenotype.
tempalphas = [0.1,0.2,0.3,0.4,0.5,0.6,0.7,0.8,0.9]
l1weights = [0.1,0.2,0.3,0.4,0.5,0.6,0.7,0.8,0.9]
tempalphas = [0.1]
l1weights = [0.1]
# The following transformation is required by Logit.
phenotype_train["Phenotype"] = phenotype_train["Phenotype"].replace({1: 0, 2: 1})
phenotype_test["Phenotype"] = phenotype_test["Phenotype"].replace({1: 0, 2: 1})
for tempalpha in tempalphas:
for l1weight in l1weights:
try:
# Fit the null model.
null_model = sm.Logit(phenotype_train["Phenotype"], sm.add_constant(covandpcs_train.iloc[:, 2:])).fit_regularized(alpha=tempalpha, L1_wt=l1weight)
#null_model = sm.Logit(phenotype_train["Phenotype"], sm.add_constant(covandpcs_train.iloc[:, 2:])).fit()
except:
print("XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX")
continue
# Prediction using null model on training data.
train_null_predicted = null_model.predict(sm.add_constant(covandpcs_train.iloc[:, 2:]))
from sklearn.metrics import roc_auc_score, confusion_matrix
from sklearn.metrics import r2_score
# Prediction using null model on test data.
test_null_predicted = null_model.predict(sm.add_constant(covandpcs_test.iloc[:, 2:]))
global prs_result
for i in threshold_values:
try:
# Get the PRS for a specific range.
prs_train = pd.read_table(traindirec+os.sep+Name+os.sep+"train_data.pv_"+f"{i}.profile", sep="\s+", usecols=["FID", "IID", "SCORE"])
except:
continue
prs_train['FID'] = prs_train['FID'].astype(str)
prs_train['IID'] = prs_train['IID'].astype(str)
try:
prs_test = pd.read_table(traindirec+os.sep+Name+os.sep+"test_data.pv_"+f"{i}.profile", sep="\s+", usecols=["FID", "IID", "SCORE"])
except:
continue
prs_test['FID'] = prs_test['FID'].astype(str)
prs_test['IID'] = prs_test['IID'].astype(str)
# Append PRS with covaraites and PCA for both datasets.
pheno_prs_train = pd.merge(covandpcs_train, prs_train, on=["FID", "IID"])
pheno_prs_test = pd.merge(covandpcs_test, prs_test, on=["FID", "IID"])
try:
# Fit the model.
model = sm.Logit(phenotype_train["Phenotype"], sm.add_constant(pheno_prs_train.iloc[:, 2:])).fit_regularized(alpha=tempalpha, L1_wt=l1weight)
#model = sm.Logit(phenotype_train["Phenotype"], sm.add_constant(pheno_prs_train.iloc[:, 2:])).fit()
except:
continue
train_best_predicted = model.predict(sm.add_constant(pheno_prs_train.iloc[:, 2:]))
test_best_predicted = model.predict(sm.add_constant(pheno_prs_test.iloc[:, 2:]))
from sklearn.metrics import roc_auc_score, confusion_matrix
# Save the information for all hyperparameters.
prs_result = prs_result._append({
"clump_p1": c1_val,
"clump_r2": c2_val,
"clump_kb": c3_val,
"p_window_size": p1_val,
"p_slide_size": p2_val,
"p_LD_threshold": p3_val,
"pvalue": i,
"numberofpca":p,
"tempalpha":str(tempalpha),
"l1weight":str(l1weight),
# Here one can use the different evaluation metrix.
# Same goes for continous phenotype in the following function.
"Train_pure_prs":roc_auc_score(phenotype_train["Phenotype"].values,prs_train['SCORE'].values),
"Train_null_model":roc_auc_score(phenotype_train["Phenotype"].values,train_null_predicted.values),
"Train_best_model":roc_auc_score(phenotype_train["Phenotype"].values,train_best_predicted.values),
"Test_pure_prs":roc_auc_score(phenotype_test["Phenotype"].values,prs_test['SCORE'].values),
"Test_null_model":roc_auc_score(phenotype_test["Phenotype"].values,test_null_predicted.values),
"Test_best_model":roc_auc_score(phenotype_test["Phenotype"].values,test_best_predicted.values),
}, ignore_index=True)
prs_result.to_csv(traindirec+os.sep+Name+os.sep+"Results.csv",index=False)
return
# This function used for prediction of continous phenotypes.
def fit_continous_phenotype_on_PRS(traindirec, newtrainfilename,p, p1_val, p2_val, p3_val, c1_val, c2_val, c3_val,Name,pvaluefile):
threshold_values = allpvalues
# Merge the covariates, pca and phenotypes.
tempphenotype_train = pd.read_table(traindirec+os.sep+newtrainfilename+".clumped.pruned"+".fam", sep="\s+",header=None)
phenotype_train = pd.DataFrame()
phenotype_train["Phenotype"] = tempphenotype_train[5].values
pcs_train = pd.read_table(traindirec+os.sep+trainfilename+".eigenvec", sep="\s+",header=None, names=["FID", "IID"] + [f"PC{str(i)}" for i in range(1, int(p)+1)])
covariate_train = pd.read_table(traindirec+os.sep+trainfilename+".cov",sep="\s+")
covariate_train.fillna(0, inplace=True)
covariate_train = covariate_train[covariate_train["FID"].isin(pcs_train["FID"].values) & covariate_train["IID"].isin(pcs_train["IID"].values)]
covariate_train['FID'] = covariate_train['FID'].astype(str)
pcs_train['FID'] = pcs_train['FID'].astype(str)
covariate_train['IID'] = covariate_train['IID'].astype(str)
pcs_train['IID'] = pcs_train['IID'].astype(str)
covandpcs_train = pd.merge(covariate_train, pcs_train, on=["FID","IID"])
covandpcs_train.fillna(0, inplace=True)
## Scale the covariates!
from sklearn.preprocessing import MinMaxScaler
from sklearn.metrics import explained_variance_score
scaler = MinMaxScaler()
normalized_values_train = scaler.fit_transform(covandpcs_train.iloc[:, 2:])
#covandpcs_train.iloc[:, 2:] = normalized_values_test
tempphenotype_test = pd.read_table(traindirec+os.sep+testfilename+".clumped.pruned"+".fam", sep="\s+",header=None)
phenotype_test= pd.DataFrame()
phenotype_test["Phenotype"] = tempphenotype_test[5].values
pcs_test = pd.read_table(traindirec+os.sep+testfilename+".eigenvec", sep="\s+",header=None, names=["FID", "IID"] + [f"PC{str(i)}" for i in range(1, int(p)+1)])
covariate_test = pd.read_table(traindirec+os.sep+testfilename+".cov",sep="\s+")
covariate_test.fillna(0, inplace=True)
covariate_test = covariate_test[covariate_test["FID"].isin(pcs_test["FID"].values) & covariate_test["IID"].isin(pcs_test["IID"].values)]
covariate_test['FID'] = covariate_test['FID'].astype(str)
pcs_test['FID'] = pcs_test['FID'].astype(str)
covariate_test['IID'] = covariate_test['IID'].astype(str)
pcs_test['IID'] = pcs_test['IID'].astype(str)
covandpcs_test = pd.merge(covariate_test, pcs_test, on=["FID","IID"])
covandpcs_test.fillna(0, inplace=True)
normalized_values_test = scaler.transform(covandpcs_test.iloc[:, 2:])
#covandpcs_test.iloc[:, 2:] = normalized_values_test
tempalphas = [0.1,0.2,0.3,0.4,0.5,0.6,0.7,0.8,0.9]
l1weights = [0.1,0.2,0.3,0.4,0.5,0.6,0.7,0.8,0.9]
tempalphas = [0.1]
l1weights = [0.1]
#phenotype_train["Phenotype"] = phenotype_train["Phenotype"].replace({1: 0, 2: 1})
#phenotype_test["Phenotype"] = phenotype_test["Phenotype"].replace({1: 0, 2: 1})
for tempalpha in tempalphas:
for l1weight in l1weights:
try:
# If explained variance is negative, use fit regualized rather than fit.
#null_model = sm.OLS(phenotype_train["Phenotype"], sm.add_constant(covandpcs_train.iloc[:, 2:])).fit_regularized(alpha=tempalpha, L1_wt=l1weight)
null_model = sm.OLS(phenotype_train["Phenotype"], sm.add_constant(covandpcs_train.iloc[:, 2:])).fit()
#null_model = sm.OLS(phenotype_train["Phenotype"], sm.add_constant(covandpcs_train.iloc[:, 2:])).fit()
except:
print("XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX")
continue
train_null_predicted = null_model.predict(sm.add_constant(covandpcs_train.iloc[:, 2:]))
from sklearn.metrics import roc_auc_score, confusion_matrix
from sklearn.metrics import r2_score
test_null_predicted = null_model.predict(sm.add_constant(covandpcs_test.iloc[:, 2:]))
global prs_result
for i in threshold_values:
try:
prs_train = pd.read_table(traindirec+os.sep+Name+os.sep+"train_data.pv_"+f"{i}.profile", sep="\s+", usecols=["FID", "IID", "SCORE"])
except:
continue
prs_train['FID'] = prs_train['FID'].astype(str)
prs_train['IID'] = prs_train['IID'].astype(str)
try:
prs_test = pd.read_table(traindirec+os.sep+Name+os.sep+"test_data.pv_"+f"{i}.profile", sep="\s+", usecols=["FID", "IID", "SCORE"])
except:
continue
prs_test['FID'] = prs_test['FID'].astype(str)
prs_test['IID'] = prs_test['IID'].astype(str)
pheno_prs_train = pd.merge(covandpcs_train, prs_train, on=["FID", "IID"])
pheno_prs_test = pd.merge(covandpcs_test, prs_test, on=["FID", "IID"])
try:
#model = sm.OLS(phenotype_train["Phenotype"], sm.add_constant(pheno_prs_train.iloc[:, 2:])).fit_regularized(alpha=tempalpha, L1_wt=l1weight)
model = sm.OLS(phenotype_train["Phenotype"], sm.add_constant(pheno_prs_train.iloc[:, 2:])).fit()
except:
continue
train_best_predicted = model.predict(sm.add_constant(pheno_prs_train.iloc[:, 2:]))
test_best_predicted = model.predict(sm.add_constant(pheno_prs_test.iloc[:, 2:]))
from sklearn.metrics import roc_auc_score, confusion_matrix
prs_result = prs_result._append({
"clump_p1": c1_val,
"clump_r2": c2_val,
"clump_kb": c3_val,
"p_window_size": p1_val,
"p_slide_size": p2_val,
"p_LD_threshold": p3_val,
"pvalue": i,
"numberofpca":p,
"tempalpha":str(tempalpha),
"l1weight":str(l1weight),
"Train_pure_prs":explained_variance_score(phenotype_train["Phenotype"],prs_train['SCORE'].values),
"Train_null_model":explained_variance_score(phenotype_train["Phenotype"],train_null_predicted),
"Train_best_model":explained_variance_score(phenotype_train["Phenotype"],train_best_predicted),
"Test_pure_prs":explained_variance_score(phenotype_test["Phenotype"],prs_test['SCORE'].values),
"Test_null_model":explained_variance_score(phenotype_test["Phenotype"],test_null_predicted),
"Test_best_model":explained_variance_score(phenotype_test["Phenotype"],test_best_predicted),
}, ignore_index=True)
prs_result.to_csv(traindirec+os.sep+Name+os.sep+"Results.csv",index=False)
return
Execute Plink#
# Define a global variable to store results
prs_result = pd.DataFrame()
def transform_plink_data(traindirec, newtrainfilename,p, p1_val, p2_val, p3_val, c1_val, c2_val, c3_val,Name,pvaluefile):
### First perform clumping on the file and save the clumpled file.
perform_clumping_and_pruning_on_individual_data(traindirec, newtrainfilename,p, p1_val, p2_val, p3_val, c1_val, c2_val, c3_val,Name,pvaluefile)
#newtrainfilename = newtrainfilename+".clumped.pruned"
#testfilename = testfilename+".clumped.pruned"
#clupmedfile = traindirec+os.sep+newtrainfilename+".clump"
#prunedfile = traindirec+os.sep+newtrainfilename+".clumped.pruned"
# Also extract the PCA at this point for both test and training data.
calculate_pca_for_traindata_testdata_for_clumped_pruned_snps(traindirec, newtrainfilename,p)
#Extract p-values from the GWAS file.
# Command for Linux.
os.system("awk "+"\'"+"{print $3,$8}"+"\'"+" ./"+filedirec+os.sep+filedirec+".txt > ./"+traindirec+os.sep+"SNP.pvalue")
# Command for windows.
### For windows get GWAK.
### https://sourceforge.net/projects/gnuwin32/
##3 Get it and place it in the same direc.
#os.system("gawk "+"\""+"{print $3,$8}"+"\""+" ./"+filedirec+os.sep+filedirec+".txt > ./"+traindirec+os.sep+"SNP.pvalue")
#print("gawk "+"\""+"{print $3,$8}"+"\""+" ./"+filedirec+os.sep+filedirec+".txt > ./"+traindirec+os.sep+"SNP.pvalue")
#exit(0)
# Caluclate Plink Score.
command = [
"./plink",
"--bfile", traindirec+os.sep+newtrainfilename+".clumped.pruned",
### SNP column = 3, Effect allele column 1 = 4, OR column=9
"--score", filedirec + os.sep +filedirec+"_Plink.txt", "3", "4", "9", "header",
"--q-score-range", traindirec+os.sep+"range_list",traindirec+os.sep+"SNP.pvalue",
"--extract", traindirec+os.sep+trainfilename+".valid.snp",
"--out", traindirec+os.sep+Name+os.sep+trainfilename
]
#exit(0)
subprocess.run(command)
command = [
"./plink",
"--bfile", folddirec+os.sep+testfilename+".clumped.pruned",
### SNP column = 3, Effect allele column 1 = 4, Beta column=12
"--score", filedirec + os.sep +filedirec+"_Plink.txt", "3", "4", "9", "header",
"--q-score-range", traindirec+os.sep+"range_list",traindirec+os.sep+"SNP.pvalue",
"--extract", traindirec+os.sep+trainfilename+".valid.snp",
"--out", folddirec+os.sep+Name+os.sep+testfilename
]
subprocess.run(command)
# At this stage the scores are finalizied.
# The next step is to fit the model
if check_phenotype_is_binary_or_continous(filedirec)=="Binary":
print("Binary Phenotype!")
fit_binary_phenotype_on_PRS(traindirec, newtrainfilename,p, p1_val, p2_val, p3_val, c1_val, c2_val, c3_val,Name,pvaluefile)
else:
print("Continous Phenotype!")
fit_continous_phenotype_on_PRS(traindirec, newtrainfilename,p, p1_val, p2_val, p3_val, c1_val, c2_val, c3_val,Name,pvaluefile)
result_directory = "Plink"
# Nested loops to iterate over different parameter values
# Create a directory to save the results.
create_directory(folddirec+os.sep+result_directory)
for p1_val in p_window_size:
for p2_val in p_slide_size:
for p3_val in p_LD_threshold:
for c1_val in clump_p1:
for c2_val in clump_r2:
for c3_val in clump_kb:
for p in numberofpca:
#pass
transform_plink_data(folddirec, newtrainfilename, p, str(p1_val), str(p2_val), str(p3_val), str(c1_val), str(c2_val), str(c3_val), result_directory, pvaluefile)
PLINK v1.90b7.2 64-bit (11 Dec 2023) www.cog-genomics.org/plink/1.9/
(C) 2005-2023 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to SampleData1/Fold_1/train_data.log.
Options in effect:
--bfile SampleData1/Fold_1/train_data.QC
--indep-pairwise 200 50 0.25
--out SampleData1/Fold_1/train_data
63761 MB RAM detected; reserving 31880 MB for main workspace.
492382 variants loaded from .bim file.
380 people (178 males, 202 females) loaded from .fam.
380 phenotype values loaded from .fam.
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 380 founders and 0 nonfounders present.
Calculating allele frequencies... 10111213141516171819202122232425262728293031323334353637383940414243444546474849505152535455565758596061626364656667686970717273747576777879808182838485868788899091929394959697989 done.
Total genotyping rate is 0.99992.
492382 variants and 380 people pass filters and QC.
Phenotype data is quantitative.
Pruned 18930 variants from chromosome 1, leaving 20356.
Pruned 19582 variants from chromosome 2, leaving 20155.
Pruned 16349 variants from chromosome 3, leaving 17137.
Pruned 15377 variants from chromosome 4, leaving 16083.
Pruned 14236 variants from chromosome 5, leaving 15375.
Pruned 19301 variants from chromosome 6, leaving 14857.
Pruned 13077 variants from chromosome 7, leaving 14043.
Pruned 12457 variants from chromosome 8, leaving 12966.
Pruned 9997 variants from chromosome 9, leaving 11471.
Pruned 11986 variants from chromosome 10, leaving 12888.
Pruned 12194 variants from chromosome 11, leaving 12229.
Pruned 10952 variants from chromosome 12, leaving 12106.
Pruned 7963 variants from chromosome 13, leaving 9213.
Pruned 7658 variants from chromosome 14, leaving 8422.
Pruned 7338 variants from chromosome 15, leaving 8205.
Pruned 8068 variants from chromosome 16, leaving 8946.
Pruned 7492 variants from chromosome 17, leaving 8386.
Pruned 6765 variants from chromosome 18, leaving 8242.
Pruned 6441 variants from chromosome 19, leaving 6447.
Pruned 5962 variants from chromosome 20, leaving 7247.
Pruned 3414 variants from chromosome 21, leaving 4116.
Pruned 3777 variants from chromosome 22, leaving 4176.
Pruning complete. 239316 of 492382 variants removed.
Marker lists written to SampleData1/Fold_1/train_data.prune.in and
SampleData1/Fold_1/train_data.prune.out .
PLINK v1.90b7.2 64-bit (11 Dec 2023) www.cog-genomics.org/plink/1.9/
(C) 2005-2023 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to SampleData1/Fold_1/train_data.log.
Options in effect:
--bfile SampleData1/Fold_1/train_data.QC
--clump SampleData1/SampleData1.txt
--clump-field P
--clump-kb 200
--clump-p1 1
--clump-r2 0.1
--clump-snp-field SNP
--extract SampleData1/Fold_1/train_data.prune.in
--out SampleData1/Fold_1/train_data
63761 MB RAM detected; reserving 31880 MB for main workspace.
492382 variants loaded from .bim file.
380 people (178 males, 202 females) loaded from .fam.
380 phenotype values loaded from .fam.
--extract: 253066 variants remaining.
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 380 founders and 0 nonfounders present.
Calculating allele frequencies... 10111213141516171819202122232425262728293031323334353637383940414243444546474849505152535455565758596061626364656667686970717273747576777879808182838485868788899091929394959697989 done.
Total genotyping rate is 0.999921.
253066 variants and 380 people pass filters and QC.
Phenotype data is quantitative.
--clump: 173148 clumps formed from 253066 top variants.
Results written to SampleData1/Fold_1/train_data.clumped .
Warning: 'rs3134762' is missing from the main dataset, and is a top variant.
Warning: 'rs3132505' is missing from the main dataset, and is a top variant.
Warning: 'rs3130424' is missing from the main dataset, and is a top variant.
246548 more top variant IDs missing; see log file.
PLINK v1.90b7.2 64-bit (11 Dec 2023) www.cog-genomics.org/plink/1.9/
(C) 2005-2023 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to SampleData1/Fold_1/train_data.QC.clumped.pruned.log.
Options in effect:
--bfile SampleData1/Fold_1/train_data.QC
--extract SampleData1/Fold_1/train_data.valid.snp
--indep-pairwise 200 50 0.25
--make-bed
--out SampleData1/Fold_1/train_data.QC.clumped.pruned
63761 MB RAM detected; reserving 31880 MB for main workspace.
492382 variants loaded from .bim file.
380 people (178 males, 202 females) loaded from .fam.
380 phenotype values loaded from .fam.
--extract: 173148 variants remaining.
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 380 founders and 0 nonfounders present.
Calculating allele frequencies... 10111213141516171819202122232425262728293031323334353637383940414243444546474849505152535455565758596061626364656667686970717273747576777879808182838485868788899091929394959697989 done.
Total genotyping rate is 0.999917.
173148 variants and 380 people pass filters and QC.
Phenotype data is quantitative.
--make-bed to SampleData1/Fold_1/train_data.QC.clumped.pruned.bed +
SampleData1/Fold_1/train_data.QC.clumped.pruned.bim +
SampleData1/Fold_1/train_data.QC.clumped.pruned.fam ... 101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960616263646566676869707172737475767778798081828384858687888990919293949596979899done.
Pruned 4 variants from chromosome 1, leaving 14074.
Pruned 3 variants from chromosome 2, leaving 13842.
Pruned 2 variants from chromosome 3, leaving 11760.
Pruned 1 variant from chromosome 4, leaving 11072.
Pruned 2 variants from chromosome 5, leaving 10631.
Pruned 50 variants from chromosome 6, leaving 10073.
Pruned 0 variants from chromosome 7, leaving 9545.
Pruned 1 variant from chromosome 8, leaving 8845.
Pruned 0 variants from chromosome 9, leaving 7761.
Pruned 7 variants from chromosome 10, leaving 8779.
Pruned 11 variants from chromosome 11, leaving 8379.
Pruned 1 variant from chromosome 12, leaving 8283.
Pruned 0 variants from chromosome 13, leaving 6342.
Pruned 0 variants from chromosome 14, leaving 5715.
Pruned 0 variants from chromosome 15, leaving 5556.
Pruned 2 variants from chromosome 16, leaving 6086.
Pruned 1 variant from chromosome 17, leaving 5753.
Pruned 0 variants from chromosome 18, leaving 5623.
Pruned 0 variants from chromosome 19, leaving 4384.
Pruned 0 variants from chromosome 20, leaving 4902.
Pruned 0 variants from chromosome 21, leaving 2825.
Pruned 0 variants from chromosome 22, leaving 2833.
Pruning complete. 85 of 173148 variants removed.
Marker lists written to
SampleData1/Fold_1/train_data.QC.clumped.pruned.prune.in and
SampleData1/Fold_1/train_data.QC.clumped.pruned.prune.out .
PLINK v1.90b7.2 64-bit (11 Dec 2023) www.cog-genomics.org/plink/1.9/
(C) 2005-2023 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to SampleData1/Fold_1/test_data.clumped.pruned.log.
Options in effect:
--bfile SampleData1/Fold_1/test_data
--extract SampleData1/Fold_1/train_data.valid.snp
--indep-pairwise 200 50 0.25
--make-bed
--out SampleData1/Fold_1/test_data.clumped.pruned
63761 MB RAM detected; reserving 31880 MB for main workspace.
551892 variants loaded from .bim file.
95 people (49 males, 46 females) loaded from .fam.
95 phenotype values loaded from .fam.
--extract: 173148 variants remaining.
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 95 founders and 0 nonfounders present.
Calculating allele frequencies... 10111213141516171819202122232425262728293031323334353637383940414243444546474849505152535455565758596061626364656667686970717273747576777879808182838485868788899091929394959697989 done.
Total genotyping rate is 0.99978.
173148 variants and 95 people pass filters and QC.
Phenotype data is quantitative.
--make-bed to SampleData1/Fold_1/test_data.clumped.pruned.bed +
SampleData1/Fold_1/test_data.clumped.pruned.bim +
SampleData1/Fold_1/test_data.clumped.pruned.fam ... 101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960616263646566676869707172737475767778798081828384858687888990919293949596979899done.
Pruned 1878 variants from chromosome 1, leaving 12200.
Pruned 1891 variants from chromosome 2, leaving 11954.
Pruned 1550 variants from chromosome 3, leaving 10212.
Pruned 1427 variants from chromosome 4, leaving 9646.
Pruned 1380 variants from chromosome 5, leaving 9253.
Pruned 1364 variants from chromosome 6, leaving 8759.
Pruned 1276 variants from chromosome 7, leaving 8269.
Pruned 1173 variants from chromosome 8, leaving 7673.
Pruned 900 variants from chromosome 9, leaving 6861.
Pruned 1118 variants from chromosome 10, leaving 7668.
Pruned 1115 variants from chromosome 11, leaving 7275.
Pruned 1022 variants from chromosome 12, leaving 7262.
Pruned 829 variants from chromosome 13, leaving 5513.
Pruned 748 variants from chromosome 14, leaving 4967.
Pruned 649 variants from chromosome 15, leaving 4907.
Pruned 697 variants from chromosome 16, leaving 5391.
Pruned 652 variants from chromosome 17, leaving 5102.
Pruned 684 variants from chromosome 18, leaving 4939.
Pruned 406 variants from chromosome 19, leaving 3978.
Pruned 577 variants from chromosome 20, leaving 4325.
Pruned 325 variants from chromosome 21, leaving 2500.
Pruned 306 variants from chromosome 22, leaving 2527.
Pruning complete. 21967 of 173148 variants removed.
Marker lists written to SampleData1/Fold_1/test_data.clumped.pruned.prune.in
and SampleData1/Fold_1/test_data.clumped.pruned.prune.out .
PLINK v1.90b7.2 64-bit (11 Dec 2023) www.cog-genomics.org/plink/1.9/
(C) 2005-2023 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to SampleData1/Fold_1/test_data.log.
Options in effect:
--bfile SampleData1/Fold_1/test_data.clumped.pruned
--extract SampleData1/Fold_1/train_data.valid.snp
--out SampleData1/Fold_1/test_data
--pca 6
63761 MB RAM detected; reserving 31880 MB for main workspace.
173148 variants loaded from .bim file.
95 people (49 males, 46 females) loaded from .fam.
95 phenotype values loaded from .fam.
--extract: 173148 variants remaining.
Using up to 8 threads (change this with --threads).
Before main variant filters, 95 founders and 0 nonfounders present.
Calculating allele frequencies... 10111213141516171819202122232425262728293031323334353637383940414243444546474849505152535455565758596061626364656667686970717273747576777879808182838485868788899091929394959697989 done.
Total genotyping rate is 0.99978.
173148 variants and 95 people pass filters and QC.
Phenotype data is quantitative.
Relationship matrix calculation complete.
--pca: Results saved to SampleData1/Fold_1/test_data.eigenval and
SampleData1/Fold_1/test_data.eigenvec .
PLINK v1.90b7.2 64-bit (11 Dec 2023) www.cog-genomics.org/plink/1.9/
(C) 2005-2023 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to SampleData1/Fold_1/train_data.log.
Options in effect:
--bfile SampleData1/Fold_1/train_data.QC.clumped.pruned
--extract SampleData1/Fold_1/train_data.valid.snp
--out SampleData1/Fold_1/train_data
--pca 6
63761 MB RAM detected; reserving 31880 MB for main workspace.
173148 variants loaded from .bim file.
380 people (178 males, 202 females) loaded from .fam.
380 phenotype values loaded from .fam.
--extract: 173148 variants remaining.
Using up to 8 threads (change this with --threads).
Before main variant filters, 380 founders and 0 nonfounders present.
Calculating allele frequencies... 10111213141516171819202122232425262728293031323334353637383940414243444546474849505152535455565758596061626364656667686970717273747576777879808182838485868788899091929394959697989 done.
Total genotyping rate is 0.999917.
173148 variants and 380 people pass filters and QC.
Phenotype data is quantitative.
Relationship matrix calculation complete.
--pca: Results saved to SampleData1/Fold_1/train_data.eigenval and
SampleData1/Fold_1/train_data.eigenvec .
PLINK v1.90b7.2 64-bit (11 Dec 2023) www.cog-genomics.org/plink/1.9/
(C) 2005-2023 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to SampleData1/Fold_1/Plink/train_data.log.
Options in effect:
--bfile SampleData1/Fold_1/train_data.QC.clumped.pruned
--extract SampleData1/Fold_1/train_data.valid.snp
--out SampleData1/Fold_1/Plink/train_data
--q-score-range SampleData1/Fold_1/range_list SampleData1/Fold_1/SNP.pvalue
--score SampleData1/SampleData1_Plink.txt 3 4 9 header
63761 MB RAM detected; reserving 31880 MB for main workspace.
173148 variants loaded from .bim file.
380 people (178 males, 202 females) loaded from .fam.
380 phenotype values loaded from .fam.
--extract: 173148 variants remaining.
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 380 founders and 0 nonfounders present.
Calculating allele frequencies... 10111213141516171819202122232425262728293031323334353637383940414243444546474849505152535455565758596061626364656667686970717273747576777879808182838485868788899091929394959697989 done.
Total genotyping rate is 0.999917.
173148 variants and 380 people pass filters and QC.
Phenotype data is quantitative.
Warning: 326469 lines skipped in --score file (326469 due to variant ID
mismatch, 0 due to allele code mismatch); see
SampleData1/Fold_1/Plink/train_data.nopred for details.
Warning: 326470 lines skipped in --q-score-range data file.
--score: 173148 valid predictors loaded.
--score: 100 ranges processed.
Results written to SampleData1/Fold_1/Plink/train_data.*.profile.
PLINK v1.90b7.2 64-bit (11 Dec 2023) www.cog-genomics.org/plink/1.9/
(C) 2005-2023 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to SampleData1/Fold_1/Plink/test_data.log.
Options in effect:
--bfile SampleData1/Fold_1/test_data.clumped.pruned
--extract SampleData1/Fold_1/train_data.valid.snp
--out SampleData1/Fold_1/Plink/test_data
--q-score-range SampleData1/Fold_1/range_list SampleData1/Fold_1/SNP.pvalue
--score SampleData1/SampleData1_Plink.txt 3 4 9 header
63761 MB RAM detected; reserving 31880 MB for main workspace.
173148 variants loaded from .bim file.
95 people (49 males, 46 females) loaded from .fam.
95 phenotype values loaded from .fam.
--extract: 173148 variants remaining.
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 95 founders and 0 nonfounders present.
Calculating allele frequencies... 10111213141516171819202122232425262728293031323334353637383940414243444546474849505152535455565758596061626364656667686970717273747576777879808182838485868788899091929394959697989 done.
Total genotyping rate is 0.99978.
173148 variants and 95 people pass filters and QC.
Phenotype data is quantitative.
Warning: 326469 lines skipped in --score file (326469 due to variant ID
mismatch, 0 due to allele code mismatch); see
SampleData1/Fold_1/Plink/test_data.nopred for details.
Warning: 326470 lines skipped in --q-score-range data file.
--score: 173148 valid predictors loaded.
--score: 100 ranges processed.
Results written to SampleData1/Fold_1/Plink/test_data.*.profile.
Continous Phenotype!
Repeat the process for each fold.#
Change the foldnumber variable.
#foldnumber = sys.argv[1]
foldnumber = "0" # Setting 'foldnumber' to "0"
Or uncomment the following line:
# foldnumber = sys.argv[1]
python Plink.py 0
python Plink.py 1
python Plink.py 2
python Plink.py 3
python Plink.py 4
The following files should exist after the execution:
SampleData1/Fold_0/Plink/Results.csvSampleData1/Fold_1/Plink/Results.csvSampleData1/Fold_2/Plink/Results.csvSampleData1/Fold_3/Plink/Results.csvSampleData1/Fold_4/Plink/Results.csv
Check the results file for each fold.#
import os
# List of file names to check for existence
f = [
"./"+filedirec+"/Fold_0"+os.sep+result_directory+"Results.csv",
"./"+filedirec+"/Fold_1"+os.sep+result_directory+"Results.csv",
"./"+filedirec+"/Fold_2"+os.sep+result_directory+"Results.csv",
"./"+filedirec+"/Fold_3"+os.sep+result_directory+"Results.csv",
"./"+filedirec+"/Fold_4"+os.sep+result_directory+"Results.csv",
]
# Loop through each file name in the list
for loop in range(0,5):
# Check if the file exists in the specified directory for the given fold
if os.path.exists(filedirec+os.sep+"Fold_"+str(loop)+os.sep+result_directory+os.sep+"Results.csv"):
temp = pd.read_csv(filedirec+os.sep+"Fold_"+str(loop)+os.sep+result_directory+os.sep+"Results.csv")
print("Fold_",loop, "Yes, the file exists.")
#print(temp.head())
print("Number of P-values processed: ",len(temp))
# Print a message indicating that the file exists
else:
# Print a message indicating that the file does not exist
print("Fold_",loop, "No, the file does not exist.")
Fold_ 0 Yes, the file exists.
Number of P-values processed: 100
Fold_ 1 Yes, the file exists.
Number of P-values processed: 100
Fold_ 2 Yes, the file exists.
Number of P-values processed: 100
Fold_ 3 Yes, the file exists.
Number of P-values processed: 100
Fold_ 4 Yes, the file exists.
Number of P-values processed: 100
Sum the results for each fold.#
print("We have to ensure when we sum the entries across all Folds, the same rows are merged!")
def sum_and_average_columns(data_frames):
"""Sum and average numerical columns across multiple DataFrames, and keep non-numerical columns unchanged."""
# Initialize DataFrame to store the summed results for numerical columns
summed_df = pd.DataFrame()
non_numerical_df = pd.DataFrame()
for df in data_frames:
# Identify numerical and non-numerical columns
numerical_cols = df.select_dtypes(include=[np.number]).columns
non_numerical_cols = df.select_dtypes(exclude=[np.number]).columns
# Sum numerical columns
if summed_df.empty:
summed_df = pd.DataFrame(0, index=range(len(df)), columns=numerical_cols)
summed_df[numerical_cols] = summed_df[numerical_cols].add(df[numerical_cols], fill_value=0)
# Keep non-numerical columns (take the first non-numerical entry for each column)
if non_numerical_df.empty:
non_numerical_df = df[non_numerical_cols]
else:
non_numerical_df[non_numerical_cols] = non_numerical_df[non_numerical_cols].combine_first(df[non_numerical_cols])
# Divide the summed values by the number of dataframes to get the average
averaged_df = summed_df / len(data_frames)
# Combine numerical and non-numerical DataFrames
result_df = pd.concat([averaged_df, non_numerical_df], axis=1)
return result_df
from functools import reduce
import os
import pandas as pd
from functools import reduce
def find_common_rows(allfoldsframe):
# Define the performance columns that need to be excluded
performance_columns = [
'Train_null_model', 'Train_pure_prs', 'Train_best_model',
'Test_pure_prs', 'Test_null_model', 'Test_best_model'
]
important_columns = [
'clump_p1',
'clump_r2',
'clump_kb',
'p_window_size',
'p_slide_size',
'p_LD_threshold',
'pvalue',
'referencepanel',
'PRSice-2_Model',
'effectsizes',
'h2model',
'model',
'numberofpca',
'tempalpha',
'l1weight',
]
# Function to remove performance columns from a DataFrame
def drop_performance_columns(df):
return df.drop(columns=performance_columns, errors='ignore')
def get_important_columns(df ):
existing_columns = [col for col in important_columns if col in df.columns]
if existing_columns:
return df[existing_columns].copy()
else:
return pd.DataFrame()
# Drop performance columns from all DataFrames in the list
allfoldsframe_dropped = [drop_performance_columns(df) for df in allfoldsframe]
# Get the important columns.
allfoldsframe_dropped = [get_important_columns(df) for df in allfoldsframe_dropped]
# Iteratively find common rows and track unique and common rows
common_rows = allfoldsframe_dropped[0]
for i in range(1, len(allfoldsframe_dropped)):
# Get the next DataFrame
next_df = allfoldsframe_dropped[i]
# Count unique rows in the current DataFrame and the next DataFrame
unique_in_common = common_rows.shape[0]
unique_in_next = next_df.shape[0]
# Find common rows between the current common_rows and the next DataFrame
common_rows = pd.merge(common_rows, next_df, how='inner')
# Count the common rows after merging
common_count = common_rows.shape[0]
# Print the unique and common row counts
print(f"Iteration {i}:")
print(f"Unique rows in current common DataFrame: {unique_in_common}")
print(f"Unique rows in next DataFrame: {unique_in_next}")
print(f"Common rows after merge: {common_count}\n")
# Now that we have the common rows, extract these from the original DataFrames
extracted_common_rows_frames = []
for original_df in allfoldsframe:
# Merge the common rows with the original DataFrame, keeping only the rows that match the common rows
extracted_common_rows = pd.merge(common_rows, original_df, how='inner', on=common_rows.columns.tolist())
# Add the DataFrame with the extracted common rows to the list
extracted_common_rows_frames.append(extracted_common_rows)
# Print the number of rows in the common DataFrames
for i, df in enumerate(extracted_common_rows_frames):
print(f"DataFrame {i + 1} with extracted common rows has {df.shape[0]} rows.")
# Return the list of DataFrames with extracted common rows
return extracted_common_rows_frames
# Example usage (assuming allfoldsframe is populated as shown earlier):
allfoldsframe = []
# Loop through each file name in the list
for loop in range(0, 5):
# Check if the file exists in the specified directory for the given fold
file_path = os.path.join(filedirec, "Fold_" + str(loop), result_directory, "Results.csv")
if os.path.exists(file_path):
allfoldsframe.append(pd.read_csv(file_path))
# Print a message indicating that the file exists
print("Fold_", loop, "Yes, the file exists.")
else:
# Print a message indicating that the file does not exist
print("Fold_", loop, "No, the file does not exist.")
# Find the common rows across all folds and return the list of extracted common rows
extracted_common_rows_list = find_common_rows(allfoldsframe)
# Sum the values column-wise
# For string values, do not sum it the values are going to be the same for each fold.
# Only sum the numeric values.
divided_result = sum_and_average_columns(extracted_common_rows_list)
print(divided_result)
We have to ensure when we sum the entries across all Folds, the same rows are merged!
Fold_ 0 Yes, the file exists.
Fold_ 1 Yes, the file exists.
Fold_ 2 Yes, the file exists.
Fold_ 3 Yes, the file exists.
Fold_ 4 Yes, the file exists.
Iteration 1:
Unique rows in current common DataFrame: 100
Unique rows in next DataFrame: 100
Common rows after merge: 100
Iteration 2:
Unique rows in current common DataFrame: 100
Unique rows in next DataFrame: 100
Common rows after merge: 100
Iteration 3:
Unique rows in current common DataFrame: 100
Unique rows in next DataFrame: 100
Common rows after merge: 100
Iteration 4:
Unique rows in current common DataFrame: 100
Unique rows in next DataFrame: 100
Common rows after merge: 100
DataFrame 1 with extracted common rows has 100 rows.
DataFrame 2 with extracted common rows has 100 rows.
DataFrame 3 with extracted common rows has 100 rows.
DataFrame 4 with extracted common rows has 100 rows.
DataFrame 5 with extracted common rows has 100 rows.
clump_p1 clump_r2 clump_kb p_window_size p_slide_size p_LD_threshold \
0 1.0 0.1 200.0 200.0 50.0 0.25
1 1.0 0.1 200.0 200.0 50.0 0.25
2 1.0 0.1 200.0 200.0 50.0 0.25
3 1.0 0.1 200.0 200.0 50.0 0.25
4 1.0 0.1 200.0 200.0 50.0 0.25
.. ... ... ... ... ... ...
95 1.0 0.1 200.0 200.0 50.0 0.25
96 1.0 0.1 200.0 200.0 50.0 0.25
97 1.0 0.1 200.0 200.0 50.0 0.25
98 1.0 0.1 200.0 200.0 50.0 0.25
99 1.0 0.1 200.0 200.0 50.0 0.25
pvalue numberofpca tempalpha l1weight Train_pure_prs \
0 1.000000e-10 6.0 0.1 0.1 0.000110
1 1.261857e-10 6.0 0.1 0.1 0.000109
2 1.592283e-10 6.0 0.1 0.1 0.000105
3 2.009233e-10 6.0 0.1 0.1 0.000104
4 2.535364e-10 6.0 0.1 0.1 0.000105
.. ... ... ... ... ...
95 3.944206e-01 6.0 0.1 0.1 0.000006
96 4.977024e-01 6.0 0.1 0.1 0.000005
97 6.280291e-01 6.0 0.1 0.1 0.000005
98 7.924829e-01 6.0 0.1 0.1 0.000004
99 1.000000e+00 6.0 0.1 0.1 0.000004
Train_null_model Train_best_model Test_pure_prs Test_null_model \
0 0.23001 0.239210 0.000103 0.118692
1 0.23001 0.239147 0.000102 0.118692
2 0.23001 0.238915 0.000096 0.118692
3 0.23001 0.239129 0.000094 0.118692
4 0.23001 0.239649 0.000096 0.118692
.. ... ... ... ...
95 0.23001 0.391233 0.000006 0.118692
96 0.23001 0.392602 0.000006 0.118692
97 0.23001 0.389123 0.000005 0.118692
98 0.23001 0.389989 0.000004 0.118692
99 0.23001 0.390565 0.000004 0.118692
Test_best_model
0 0.115508
1 0.114823
2 0.114201
3 0.115284
4 0.115394
.. ...
95 0.330556
96 0.331492
97 0.328775
98 0.333163
99 0.333190
[100 rows x 16 columns]
Results#
1. Reporting Based on Best Training Performance:#
One can report the results based on the best performance of the training data. For example, if for a specific combination of hyperparameters, the training performance is high, report the corresponding test performance.
Example code:
df = divided_result.sort_values(by='Train_best_model', ascending=False) print(df.iloc[0].to_markdown())
Binary Phenotypes Result Analysis#
You can find the performance quality for binary phenotype using the following template:
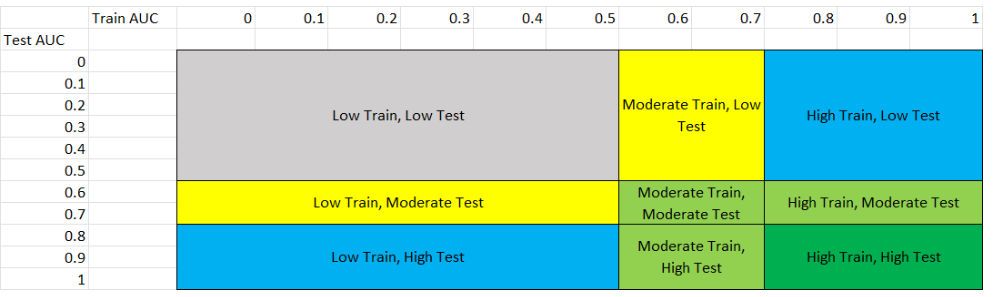
This figure shows the 8 different scenarios that can exist in the results, and the following table explains each scenario.
We classified performance based on the following table:
Performance Level |
Range |
|---|---|
Low Performance |
0 to 0.5 |
Moderate Performance |
0.6 to 0.7 |
High Performance |
0.8 to 1 |
You can match the performance based on the following scenarios:
Scenario |
What’s Happening |
Implication |
|---|---|---|
High Test, High Train |
The model performs well on both training and test datasets, effectively learning the underlying patterns. |
The model is well-tuned, generalizes well, and makes accurate predictions on both datasets. |
High Test, Moderate Train |
The model generalizes well but may not be fully optimized on training data, missing some underlying patterns. |
The model is fairly robust but may benefit from further tuning or more training to improve its learning. |
High Test, Low Train |
An unusual scenario, potentially indicating data leakage or overestimation of test performance. |
The model’s performance is likely unreliable; investigate potential data issues or random noise. |
Moderate Test, High Train |
The model fits the training data well but doesn’t generalize as effectively, capturing only some test patterns. |
The model is slightly overfitting; adjustments may be needed to improve generalization on unseen data. |
Moderate Test, Moderate Train |
The model shows balanced but moderate performance on both datasets, capturing some patterns but missing others. |
The model is moderately fitting; further improvements could be made in both training and generalization. |
Moderate Test, Low Train |
The model underperforms on training data and doesn’t generalize well, leading to moderate test performance. |
The model may need more complexity, additional features, or better training to improve on both datasets. |
Low Test, High Train |
The model overfits the training data, performing poorly on the test set. |
The model doesn’t generalize well; simplifying the model or using regularization may help reduce overfitting. |
Low Test, Low Train |
The model performs poorly on both training and test datasets, failing to learn the data patterns effectively. |
The model is underfitting; it may need more complexity, additional features, or more data to improve performance. |
Recommendations for Publishing Results#
When publishing results, scenarios with moderate train and moderate test performance can be used for complex phenotypes or diseases. However, results showing high train and moderate test, high train and high test, and moderate train and high test are recommended.
For most phenotypes, results typically fall in the moderate train and moderate test performance category.
Continuous Phenotypes Result Analysis#
You can find the performance quality for continuous phenotypes using the following template:
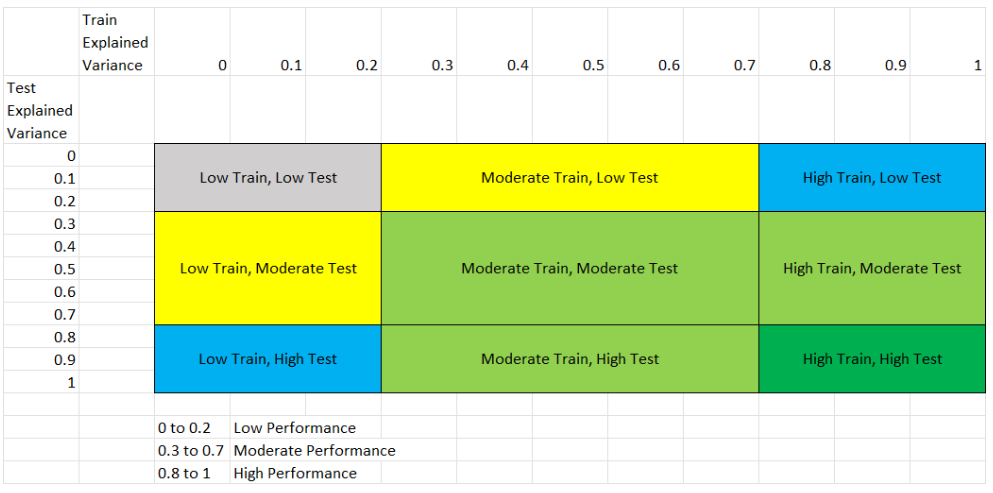
This figure shows the 8 different scenarios that can exist in the results, and the following table explains each scenario.
We classified performance based on the following table:
Performance Level |
Range |
|---|---|
Low Performance |
0 to 0.2 |
Moderate Performance |
0.3 to 0.7 |
High Performance |
0.8 to 1 |
You can match the performance based on the following scenarios:
Scenario |
What’s Happening |
Implication |
|---|---|---|
High Test, High Train |
The model performs well on both training and test datasets, effectively learning the underlying patterns. |
The model is well-tuned, generalizes well, and makes accurate predictions on both datasets. |
High Test, Moderate Train |
The model generalizes well but may not be fully optimized on training data, missing some underlying patterns. |
The model is fairly robust but may benefit from further tuning or more training to improve its learning. |
High Test, Low Train |
An unusual scenario, potentially indicating data leakage or overestimation of test performance. |
The model’s performance is likely unreliable; investigate potential data issues or random noise. |
Moderate Test, High Train |
The model fits the training data well but doesn’t generalize as effectively, capturing only some test patterns. |
The model is slightly overfitting; adjustments may be needed to improve generalization on unseen data. |
Moderate Test, Moderate Train |
The model shows balanced but moderate performance on both datasets, capturing some patterns but missing others. |
The model is moderately fitting; further improvements could be made in both training and generalization. |
Moderate Test, Low Train |
The model underperforms on training data and doesn’t generalize well, leading to moderate test performance. |
The model may need more complexity, additional features, or better training to improve on both datasets. |
Low Test, High Train |
The model overfits the training data, performing poorly on the test set. |
The model doesn’t generalize well; simplifying the model or using regularization may help reduce overfitting. |
Low Test, Low Train |
The model performs poorly on both training and test datasets, failing to learn the data patterns effectively. |
The model is underfitting; it may need more complexity, additional features, or more data to improve performance. |
Recommendations for Publishing Results#
When publishing results, scenarios with moderate train and moderate test performance can be used for complex phenotypes or diseases. However, results showing high train and moderate test, high train and high test, and moderate train and high test are recommended.
For most continuous phenotypes, results typically fall in the moderate train and moderate test performance category.
2. Reporting Generalized Performance:#
One can also report the generalized performance by calculating the difference between the training and test performance, and the sum of the test and training performance. Report the result or hyperparameter combination for which the sum is high and the difference is minimal.
Example code:
df = divided_result.copy() df['Difference'] = abs(df['Train_best_model'] - df['Test_best_model']) df['Sum'] = df['Train_best_model'] + df['Test_best_model'] sorted_df = df.sort_values(by=['Sum', 'Difference'], ascending=[False, True]) print(sorted_df.iloc[0].to_markdown())
3. Reporting Hyperparameters Affecting Test and Train Performance:#
Find the hyperparameters that have more than one unique value and calculate their correlation with the following columns to understand how they are affecting the performance of train and test sets:
Train_null_modelTrain_pure_prsTrain_best_modelTest_pure_prsTest_null_modelTest_best_model
4. Other Analysis#
Once you have the results, you can find how hyperparameters affect the model performance.
Analysis, like overfitting and underfitting, can be performed as well.
The way you are going to report the results can vary.
Results can be visualized, and other patterns in the data can be explored.
import pandas as pd
import seaborn as sns
import matplotlib.pyplot as plt
%matplotlib notebook
import matplotlib
import numpy as np
import matplotlib.pyplot as plt
df = divided_result.sort_values(by='Train_best_model', ascending=False)
print("1. Reporting Based on Best Training Performance:\n")
print(df.iloc[0].to_markdown())
df = divided_result.copy()
# Plot Train and Test best models against p-values
plt.figure(figsize=(10, 6))
plt.plot(df['pvalue'], df['Train_best_model'], label='Train_best_model', marker='o', color='royalblue')
plt.plot(df['pvalue'], df['Test_best_model'], label='Test_best_model', marker='o', color='darkorange')
# Highlight the p-value where both train and test are high
best_index = df[['Train_best_model']].sum(axis=1).idxmax()
best_pvalue = df.loc[best_index, 'pvalue']
best_train = df.loc[best_index, 'Train_best_model']
best_test = df.loc[best_index, 'Test_best_model']
# Use dark colors for the circles
plt.scatter(best_pvalue, best_train, color='darkred', s=100, label=f'Best Performance (Train)', edgecolor='black', zorder=5)
plt.scatter(best_pvalue, best_test, color='darkblue', s=100, label=f'Best Performance (Test)', edgecolor='black', zorder=5)
# Annotate the best performance with p-value, train, and test values
plt.text(best_pvalue, best_train, f'p={best_pvalue:.4g}\nTrain={best_train:.4g}', ha='right', va='bottom', fontsize=9, color='darkred')
plt.text(best_pvalue, best_test, f'p={best_pvalue:.4g}\nTest={best_test:.4g}', ha='right', va='top', fontsize=9, color='darkblue')
# Calculate Difference and Sum
df['Difference'] = abs(df['Train_best_model'] - df['Test_best_model'])
df['Sum'] = df['Train_best_model'] + df['Test_best_model']
# Sort the DataFrame
sorted_df = df.sort_values(by=['Sum', 'Difference'], ascending=[False, True])
#sorted_df = df.sort_values(by=[ 'Difference','Sum'], ascending=[ True,False])
# Highlight the general performance
general_index = sorted_df.index[0]
general_pvalue = sorted_df.loc[general_index, 'pvalue']
general_train = sorted_df.loc[general_index, 'Train_best_model']
general_test = sorted_df.loc[general_index, 'Test_best_model']
plt.scatter(general_pvalue, general_train, color='darkgreen', s=150, label='General Performance (Train)', edgecolor='black', zorder=6)
plt.scatter(general_pvalue, general_test, color='darkorange', s=150, label='General Performance (Test)', edgecolor='black', zorder=6)
# Annotate the general performance with p-value, train, and test values
plt.text(general_pvalue, general_train, f'p={general_pvalue:.4g}\nTrain={general_train:.4g}', ha='left', va='bottom', fontsize=9, color='darkgreen')
plt.text(general_pvalue, general_test, f'p={general_pvalue:.4g}\nTest={general_test:.4g}', ha='left', va='top', fontsize=9, color='darkorange')
# Add labels and legend
plt.xlabel('p-value')
plt.ylabel('Model Performance')
plt.title('Train vs Test Best Models')
plt.legend()
plt.show()
print("2. Reporting Generalized Performance:\n")
df = divided_result.copy()
df['Difference'] = abs(df['Train_best_model'] - df['Test_best_model'])
df['Sum'] = df['Train_best_model'] + df['Test_best_model']
sorted_df = df.sort_values(by=['Sum', 'Difference'], ascending=[False, True])
print(sorted_df.iloc[0].to_markdown())
print("3. Reporting the correlation of hyperparameters and the performance of 'Train_null_model', 'Train_pure_prs', 'Train_best_model', 'Test_pure_prs', 'Test_null_model', and 'Test_best_model':\n")
print("3. For string hyperparameters, we used one-hot encoding to find the correlation between string hyperparameters and 'Train_null_model', 'Train_pure_prs', 'Train_best_model', 'Test_pure_prs', 'Test_null_model', and 'Test_best_model'.")
print("3. We performed this analysis for those hyperparameters that have more than one unique value.")
correlation_columns = [
'Train_null_model', 'Train_pure_prs', 'Train_best_model',
'Test_pure_prs', 'Test_null_model', 'Test_best_model'
]
hyperparams = [col for col in divided_result.columns if len(divided_result[col].unique()) > 1]
hyperparams = list(set(hyperparams+correlation_columns))
# Separate numeric and string columns
numeric_hyperparams = [col for col in hyperparams if pd.api.types.is_numeric_dtype(divided_result[col])]
string_hyperparams = [col for col in hyperparams if pd.api.types.is_string_dtype(divided_result[col])]
# Encode string columns using one-hot encoding
divided_result_encoded = pd.get_dummies(divided_result, columns=string_hyperparams)
# Combine numeric hyperparams with the new one-hot encoded columns
encoded_columns = [col for col in divided_result_encoded.columns if col.startswith(tuple(string_hyperparams))]
hyperparams = numeric_hyperparams + encoded_columns
# Calculate correlations
correlations = divided_result_encoded[hyperparams].corr()
# Display correlation of hyperparameters with train/test performance columns
hyperparam_correlations = correlations.loc[hyperparams, correlation_columns]
hyperparam_correlations = hyperparam_correlations.fillna(0)
# Plotting the correlation heatmap
plt.figure(figsize=(12, 8))
ax = sns.heatmap(hyperparam_correlations, annot=True, cmap='viridis', fmt='.2f', cbar=True)
ax.set_xticklabels(ax.get_xticklabels(), rotation=90, ha='right')
# Rotate y-axis labels to horizontal
#ax.set_yticklabels(ax.get_yticklabels(), rotation=0, va='center')
plt.title('Correlation of Hyperparameters with Train/Test Performance')
plt.show()
sns.set_theme(style="whitegrid") # Choose your preferred style
pairplot = sns.pairplot(divided_result_encoded[hyperparams],hue = 'Test_best_model', palette='viridis')
# Adjust the figure size
pairplot.fig.set_size_inches(15, 15) # You can adjust the size as needed
for ax in pairplot.axes.flatten():
ax.set_xlabel(ax.get_xlabel(), rotation=90, ha='right') # X-axis labels vertical
#ax.set_ylabel(ax.get_ylabel(), rotation=0, va='bottom') # Y-axis labels horizontal
# Show the plot
plt.show()
1. Reporting Based on Best Training Performance:
| | 94 |
|:-----------------|--------------:|
| clump_p1 | 1 |
| clump_r2 | 0.1 |
| clump_kb | 200 |
| p_window_size | 200 |
| p_slide_size | 50 |
| p_LD_threshold | 0.25 |
| pvalue | 0.312572 |
| numberofpca | 6 |
| tempalpha | 0.1 |
| l1weight | 0.1 |
| Train_pure_prs | 6.53073e-06 |
| Train_null_model | 0.23001 |
| Train_best_model | 0.393524 |
| Test_pure_prs | 6.75312e-06 |
| Test_null_model | 0.118692 |
| Test_best_model | 0.340104 |

2. Reporting Generalized Performance:
| | 94 |
|:-----------------|--------------:|
| clump_p1 | 1 |
| clump_r2 | 0.1 |
| clump_kb | 200 |
| p_window_size | 200 |
| p_slide_size | 50 |
| p_LD_threshold | 0.25 |
| pvalue | 0.312572 |
| numberofpca | 6 |
| tempalpha | 0.1 |
| l1weight | 0.1 |
| Train_pure_prs | 6.53073e-06 |
| Train_null_model | 0.23001 |
| Train_best_model | 0.393524 |
| Test_pure_prs | 6.75312e-06 |
| Test_null_model | 0.118692 |
| Test_best_model | 0.340104 |
| Difference | 0.0534201 |
| Sum | 0.733628 |
3. Reporting the correlation of hyperparameters and the performance of 'Train_null_model', 'Train_pure_prs', 'Train_best_model', 'Test_pure_prs', 'Test_null_model', and 'Test_best_model':
3. For string hyperparameters, we used one-hot encoding to find the correlation between string hyperparameters and 'Train_null_model', 'Train_pure_prs', 'Train_best_model', 'Test_pure_prs', 'Test_null_model', and 'Test_best_model'.
3. We performed this analysis for those hyperparameters that have more than one unique value.

