GCTB-SBayesRC#
GCTB-SBAYESRC operates similarly to GCTB-SBAYES, but with two key additional features:
It imputes SNPs that are not present in the GWAS, thereby increasing the number of SNPs in the GWAS dataset.
It utilizes the Eigen-decomposition data of LD matrices to generate new beta values.
Reference Panels Eigen Values#
Save the downloaded frame in the folder SBayesR_LDFrames.
You can download these LD Matrices and save them in the folder SBayesR_LDFrames:
ukbEUR_HM3ukbEUR_HM3.zipukbEUR_ImputedukbEUR_Imputed.zip
Execution#
Step 1: QC & Imputation of Summary Statistics for SNPs in LD Reference but not in GWAS Data#
gctb --ldm-eigen ldm --gwas-summary test.ma --impute-summary --out test
Step 2: Run SBayesRC with Annotation Data#
gctb --ldm-eigen ldm --gwas-summary test.imputed.ma --sbayes RC --annot annot.txt --out test
GCTB Hyperparameters#
We considered the following hyperparameters from all options provided by GCTB:
LD panels:
ukbEUR_HM3andukbEUR_Imputed--gwas-summary: Path to the GWAS summary statistics in.maformat.
The above parameters can be specified in the GCTB command. Additionally, we considered the following parameters:
GWAS file processing for GCTB-SBayesRC#
When the effect size relates to disease risk and is thus given as an odds ratio (OR) rather than BETA (for continuous traits), the PRS is computed as a product of ORs. To simplify this calculation, take the natural logarithm of the OR so that the PRS can be computed using summation instead.
For continuous phenotype GWAS, the SampleData1/SampleData1.gz file should have BETAs, and for binary phenotypes, it should have OR instead of BETAs. If BETAs are not available, we convert OR to BETAs using BETA = np.log(OR) and convert BETAs to OR using OR = np.exp(BETA).
import numpy as np; np is the NumPy module.
import os
import pandas as pd
import numpy as np
import os
import pandas as pd
import numpy as np
def check_phenotype_is_binary_or_continous(filedirec):
# Read the processed quality controlled file for a phenotype
df = pd.read_csv(filedirec+os.sep+filedirec+'_QC.fam',sep="\s+",header=None)
column_values = df[5].unique()
if len(set(column_values)) == 2:
return "Binary"
else:
return "Continous"
#filedirec = sys.argv[1]
filedirec = "SampleData1"
#filedirec = "asthma_19"
#filedirec = "migraine_0"
# Read the GWAS file.
GWAS = filedirec + os.sep + filedirec+".gz"
df = pd.read_csv(GWAS,compression= "gzip",sep="\s+")
if "BETA" in df.columns.to_list():
# For Continous Phenotype.
df = df[['CHR', 'BP', 'SNP', 'A1', 'A2', 'N', 'SE', 'P', 'BETA', 'INFO', 'MAF']]
else:
df["BETA"] = np.log(df["OR"])
df = df[['CHR', 'BP', 'SNP', 'A1', 'A2', 'N', 'SE', 'P', 'BETA', 'INFO', 'MAF']]
df.to_csv(filedirec + os.sep +filedirec+".txt",sep="\t",index=False)
print(df.head().to_markdown())
print("Length of DataFrame!",len(df))
df_transformed = pd.DataFrame({
'SNP': df['SNP'],
'A1': df['A1'],
'A2': df['A2'],
'freq': df['MAF'],
'b': df['BETA'],
'se': df['SE'],
'p': df['P'],
'N': df['N']
})
# Save this file as it is being required by GCTA.
output_file = filedirec+os.sep+"SBayesR.ma"
df_transformed.to_csv(output_file,sep="\t",index=False)
print(df_transformed.head().to_markdown())
print("Length of DataFrame!",len(df_transformed))
| | CHR | BP | SNP | A1 | A2 | N | SE | P | BETA | INFO | MAF |
|---:|------:|-------:|:-----------|:-----|:-----|-------:|-----------:|---------:|------------:|---------:|---------:|
| 0 | 1 | 756604 | rs3131962 | A | G | 388028 | 0.00301666 | 0.483171 | -0.00211532 | 0.890558 | 0.36939 |
| 1 | 1 | 768448 | rs12562034 | A | G | 388028 | 0.00329472 | 0.834808 | 0.00068708 | 0.895894 | 0.336846 |
| 2 | 1 | 779322 | rs4040617 | G | A | 388028 | 0.00303344 | 0.42897 | -0.00239932 | 0.897508 | 0.377368 |
| 3 | 1 | 801536 | rs79373928 | G | T | 388028 | 0.00841324 | 0.808999 | 0.00203363 | 0.908963 | 0.483212 |
| 4 | 1 | 808631 | rs11240779 | G | A | 388028 | 0.00242821 | 0.590265 | 0.00130747 | 0.893213 | 0.45041 |
Length of DataFrame! 499617
| | SNP | A1 | A2 | freq | b | se | p | N |
|---:|:-----------|:-----|:-----|---------:|------------:|-----------:|---------:|-------:|
| 0 | rs3131962 | A | G | 0.36939 | -0.00211532 | 0.00301666 | 0.483171 | 388028 |
| 1 | rs12562034 | A | G | 0.336846 | 0.00068708 | 0.00329472 | 0.834808 | 388028 |
| 2 | rs4040617 | G | A | 0.377368 | -0.00239932 | 0.00303344 | 0.42897 | 388028 |
| 3 | rs79373928 | G | T | 0.483212 | 0.00203363 | 0.00841324 | 0.808999 | 388028 |
| 4 | rs11240779 | G | A | 0.45041 | 0.00130747 | 0.00242821 | 0.590265 | 388028 |
Length of DataFrame! 499617
Define Hyperparameters#
Define hyperparameters to be optimized and set initial values.
Extract Valid SNPs from Clumped File#
For Windows, download gwak, and for Linux, the awk command is sufficient. For Windows, GWAK is required. You can download it from here. Get it and place it in the same directory.
Execution Path#
At this stage, we have the genotype training data newtrainfilename = "train_data.QC" and genotype test data newtestfilename = "test_data.QC".
We modified the following variables:
filedirec = "SampleData1"orfiledirec = sys.argv[1]foldnumber = "0"orfoldnumber = sys.argv[2]for HPC.
Only these two variables can be modified to execute the code for specific data and specific folds. Though the code can be executed separately for each fold on HPC and separately for each dataset, it is recommended to execute it for multiple diseases and one fold at a time. Here’s the corrected text in Markdown format:
P-values#
PRS calculation relies on P-values. SNPs with low P-values, indicating a high degree of association with a specific trait, are considered for calculation.
You can modify the code below to consider a specific set of P-values and save the file in the same format.
We considered the following parameters:
Minimum P-value:
1e-10Maximum P-value:
1.0Minimum exponent:
10(Minimum P-value in exponent)Number of intervals:
100(Number of intervals to be considered)
The code generates an array of logarithmically spaced P-values:
import numpy as np
import os
minimumpvalue = 10 # Minimum exponent for P-values
numberofintervals = 100 # Number of intervals to be considered
allpvalues = np.logspace(-minimumpvalue, 0, numberofintervals, endpoint=True) # Generating an array of logarithmically spaced P-values
print("Minimum P-value:", allpvalues[0])
print("Maximum P-value:", allpvalues[-1])
count = 1
with open(os.path.join(folddirec, 'range_list'), 'w') as file:
for value in allpvalues:
file.write(f'pv_{value} 0 {value}\n') # Writing range information to the 'range_list' file
count += 1
pvaluefile = os.path.join(folddirec, 'range_list')
In this code:
minimumpvaluedefines the minimum exponent for P-values.numberofintervalsspecifies how many intervals to consider.allpvaluesgenerates an array of P-values spaced logarithmically.The script writes these P-values to a file named
range_listin the specified directory.
from operator import index
import pandas as pd
import numpy as np
import os
import subprocess
import sys
import pandas as pd
import statsmodels.api as sm
import pandas as pd
from sklearn.metrics import roc_auc_score, confusion_matrix
from statsmodels.stats.contingency_tables import mcnemar
def create_directory(directory):
"""Function to create a directory if it doesn't exist."""
if not os.path.exists(directory): # Checking if the directory doesn't exist
os.makedirs(directory) # Creating the directory if it doesn't exist
return directory # Returning the created or existing directory
#foldnumber = sys.argv[1]
foldnumber = "0" # Setting 'foldnumber' to "0"
folddirec = filedirec + os.sep + "Fold_" + foldnumber # Creating a directory path for the specific fold
trainfilename = "train_data" # Setting the name of the training data file
newtrainfilename = "train_data.QC" # Setting the name of the new training data file
testfilename = "test_data" # Setting the name of the test data file
newtestfilename = "test_data.QC" # Setting the name of the new test data file
# Number of PCA to be included as a covariate.
numberofpca = ["6"] # Setting the number of PCA components to be included
# Clumping parameters.
clump_p1 = [1] # List containing clump parameter 'p1'
clump_r2 = [0.1] # List containing clump parameter 'r2'
clump_kb = [200] # List containing clump parameter 'kb'
# Pruning parameters.
p_window_size = [200] # List containing pruning parameter 'window_size'
p_slide_size = [50] # List containing pruning parameter 'slide_size'
p_LD_threshold = [0.25] # List containing pruning parameter 'LD_threshold'
# Kindly note that the number of p-values to be considered varies, and the actual p-value depends on the dataset as well.
# We will specify the range list here.
minimumpvalue = 10 # Minimum p-value in exponent
numberofintervals = 20 # Number of intervals to be considered
allpvalues = np.logspace(-minimumpvalue, 0, numberofintervals, endpoint=True) # Generating an array of logarithmically spaced p-values
count = 1
with open(folddirec + os.sep + 'range_list', 'w') as file:
for value in allpvalues:
file.write(f'pv_{value} 0 {value}\n') # Writing range information to the 'range_list' file
count = count + 1
pvaluefile = folddirec + os.sep + 'range_list'
# Initializing an empty DataFrame with specified column names
prs_result = pd.DataFrame(columns=["clump_p1", "clump_r2", "clump_kb", "bayesmodel","p_window_size", "p_slide_size", "p_LD_threshold",
"pvalue", "model","numberofpca","h2","lambda","numberofvariants(m)","Train_pure_prs", "Train_null_model", "Train_best_model",
"Test_pure_prs", "Test_null_model", "Test_best_model"])
Define Helper Functions#
Perform Clumping and Pruning
Calculate PCA Using Plink
Fit Binary Phenotype and Save Results
Fit Continuous Phenotype and Save Results
import os
import subprocess
import pandas as pd
import statsmodels.api as sm
from sklearn.metrics import explained_variance_score
def perform_clumping_and_pruning_on_individual_data(traindirec, newtrainfilename,numberofpca, p1_val, p2_val, p3_val, c1_val, c2_val, c3_val,Name,pvaluefile):
command = [
"./plink",
"--bfile", traindirec+os.sep+newtrainfilename,
"--indep-pairwise", p1_val, p2_val, p3_val,
"--out", traindirec+os.sep+trainfilename
]
subprocess.run(command)
# First perform pruning and then clumping and the pruning.
command = [
"./plink",
"--bfile", traindirec+os.sep+newtrainfilename,
"--clump-p1", c1_val,
"--extract", traindirec+os.sep+trainfilename+".prune.in",
"--clump-r2", c2_val,
"--clump-kb", c3_val,
"--clump", filedirec+os.sep+filedirec+".txt",
"--clump-snp-field", "SNP",
"--clump-field", "P",
"--out", traindirec+os.sep+trainfilename
]
subprocess.run(command)
# Extract the valid SNPs from th clumped file.
# For windows download gwak for linux awk commmand is sufficient.
### For windows require GWAK.
### https://sourceforge.net/projects/gnuwin32/
##3 Get it and place it in the same direc.
#os.system("gawk "+"\""+"NR!=1{print $3}"+"\" "+ traindirec+os.sep+trainfilename+".clumped > "+traindirec+os.sep+trainfilename+".valid.snp")
#print("gawk "+"\""+"NR!=1{print $3}"+"\" "+ traindirec+os.sep+trainfilename+".clumped > "+traindirec+os.sep+trainfilename+".valid.snp")
#Linux:
command = f"awk 'NR!=1{{print $3}}' {traindirec}{os.sep}{trainfilename}.clumped > {traindirec}{os.sep}{trainfilename}.valid.snp"
os.system(command)
command = [
"./plink",
"--make-bed",
"--bfile", traindirec+os.sep+newtrainfilename,
"--indep-pairwise", p1_val, p2_val, p3_val,
"--extract", traindirec+os.sep+trainfilename+".valid.snp",
"--out", traindirec+os.sep+newtrainfilename+".clumped.pruned"
]
subprocess.run(command)
command = [
"./plink",
"--make-bed",
"--bfile", traindirec+os.sep+testfilename,
"--indep-pairwise", p1_val, p2_val, p3_val,
"--extract", traindirec+os.sep+trainfilename+".valid.snp",
"--out", traindirec+os.sep+testfilename+".clumped.pruned"
]
subprocess.run(command)
def calculate_pca_for_traindata_testdata_for_clumped_pruned_snps(traindirec, newtrainfilename,p):
# Calculate the PRS for the test data using the same set of SNPs and also calculate the PCA.
# Also extract the PCA at this point.
# PCA are calculated afer clumping and pruining.
command = [
"./plink",
"--bfile", folddirec+os.sep+testfilename+".clumped.pruned",
# Select the final variants after clumping and pruning.
"--extract", traindirec+os.sep+trainfilename+".valid.snp",
"--pca", p,
"--out", folddirec+os.sep+testfilename
]
subprocess.run(command)
command = [
"./plink",
"--bfile", traindirec+os.sep+newtrainfilename+".clumped.pruned",
# Select the final variants after clumping and pruning.
"--extract", traindirec+os.sep+trainfilename+".valid.snp",
"--pca", p,
"--out", traindirec+os.sep+trainfilename
]
subprocess.run(command)
# This function fit the binary model on the PRS.
def fit_binary_phenotype_on_PRS(traindirec, newtrainfilename,panel, p, p1_val, p2_val, p3_val, c1_val, c2_val, c3_val,Name,pvaluefile):
threshold_values = allpvalues
# Merge the covariates, pca and phenotypes.
tempphenotype_train = pd.read_table(traindirec+os.sep+newtrainfilename+".clumped.pruned"+".fam", sep="\s+",header=None)
phenotype_train = pd.DataFrame()
phenotype_train["Phenotype"] = tempphenotype_train[5].values
pcs_train = pd.read_table(traindirec+os.sep+trainfilename+".eigenvec", sep="\s+",header=None, names=["FID", "IID"] + [f"PC{str(i)}" for i in range(1, int(p)+1)])
covariate_train = pd.read_table(traindirec+os.sep+trainfilename+".cov",sep="\s+")
covariate_train.fillna(0, inplace=True)
covariate_train = covariate_train[covariate_train["FID"].isin(pcs_train["FID"].values) & covariate_train["IID"].isin(pcs_train["IID"].values)]
covariate_train['FID'] = covariate_train['FID'].astype(str)
pcs_train['FID'] = pcs_train['FID'].astype(str)
covariate_train['IID'] = covariate_train['IID'].astype(str)
pcs_train['IID'] = pcs_train['IID'].astype(str)
covandpcs_train = pd.merge(covariate_train, pcs_train, on=["FID","IID"])
covandpcs_train.fillna(0, inplace=True)
## Scale the covariates!
from sklearn.preprocessing import MinMaxScaler
from sklearn.metrics import explained_variance_score
scaler = MinMaxScaler()
normalized_values_train = scaler.fit_transform(covandpcs_train.iloc[:, 2:])
#covandpcs_train.iloc[:, 2:] = normalized_values_test
tempphenotype_test = pd.read_table(traindirec+os.sep+testfilename+".clumped.pruned"+".fam", sep="\s+",header=None)
phenotype_test= pd.DataFrame()
phenotype_test["Phenotype"] = tempphenotype_test[5].values
pcs_test = pd.read_table(traindirec+os.sep+testfilename+".eigenvec", sep="\s+",header=None, names=["FID", "IID"] + [f"PC{str(i)}" for i in range(1, int(p)+1)])
covariate_test = pd.read_table(traindirec+os.sep+testfilename+".cov",sep="\s+")
covariate_test.fillna(0, inplace=True)
covariate_test = covariate_test[covariate_test["FID"].isin(pcs_test["FID"].values) & covariate_test["IID"].isin(pcs_test["IID"].values)]
covariate_test['FID'] = covariate_test['FID'].astype(str)
pcs_test['FID'] = pcs_test['FID'].astype(str)
covariate_test['IID'] = covariate_test['IID'].astype(str)
pcs_test['IID'] = pcs_test['IID'].astype(str)
covandpcs_test = pd.merge(covariate_test, pcs_test, on=["FID","IID"])
covandpcs_test.fillna(0, inplace=True)
normalized_values_test = scaler.transform(covandpcs_test.iloc[:, 2:])
#covandpcs_test.iloc[:, 2:] = normalized_values_test
tempalphas = [0.1,0.2,0.3,0.4,0.5,0.6,0.7,0.8,0.9]
l1weights = [0.1,0.2,0.3,0.4,0.5,0.6,0.7,0.8,0.9]
tempalphas = [0.1]
l1weights = [0.1]
phenotype_train["Phenotype"] = phenotype_train["Phenotype"].replace({1: 0, 2: 1})
phenotype_test["Phenotype"] = phenotype_test["Phenotype"].replace({1: 0, 2: 1})
for tempalpha in tempalphas:
for l1weight in l1weights:
try:
null_model = sm.Logit(phenotype_train["Phenotype"], sm.add_constant(covandpcs_train.iloc[:, 2:])).fit_regularized(alpha=tempalpha, L1_wt=l1weight)
#null_model = sm.Logit(phenotype_train["Phenotype"], sm.add_constant(covandpcs_train.iloc[:, 2:])).fit()
except:
print("XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX")
continue
train_null_predicted = null_model.predict(sm.add_constant(covandpcs_train.iloc[:, 2:]))
from sklearn.metrics import roc_auc_score, confusion_matrix
from sklearn.metrics import r2_score
test_null_predicted = null_model.predict(sm.add_constant(covandpcs_test.iloc[:, 2:]))
global prs_result
for i in threshold_values:
try:
prs_train = pd.read_table(traindirec+os.sep+Name+os.sep+"train_data.pv_"+f"{i}.profile", sep="\s+", usecols=["FID", "IID", "SCORE"])
except:
continue
prs_train['FID'] = prs_train['FID'].astype(str)
prs_train['IID'] = prs_train['IID'].astype(str)
try:
prs_test = pd.read_table(traindirec+os.sep+Name+os.sep+"test_data.pv_"+f"{i}.profile", sep="\s+", usecols=["FID", "IID", "SCORE"])
except:
continue
prs_test['FID'] = prs_test['FID'].astype(str)
prs_test['IID'] = prs_test['IID'].astype(str)
pheno_prs_train = pd.merge(covandpcs_train, prs_train, on=["FID", "IID"])
pheno_prs_test = pd.merge(covandpcs_test, prs_test, on=["FID", "IID"])
try:
model = sm.Logit(phenotype_train["Phenotype"], sm.add_constant(pheno_prs_train.iloc[:, 2:])).fit_regularized(alpha=tempalpha, L1_wt=l1weight)
#model = sm.Logit(phenotype_train["Phenotype"], sm.add_constant(pheno_prs_train.iloc[:, 2:])).fit()
except:
continue
train_best_predicted = model.predict(sm.add_constant(pheno_prs_train.iloc[:, 2:]))
test_best_predicted = model.predict(sm.add_constant(pheno_prs_test.iloc[:, 2:]))
from sklearn.metrics import roc_auc_score, confusion_matrix
prs_result = prs_result._append({
"clump_p1": c1_val,
"clump_r2": c2_val,
"clump_kb": c3_val,
"p_window_size": p1_val,
"p_slide_size": p2_val,
"p_LD_threshold": p3_val,
"pvalue": i,
"numberofpca":p,
"tempalpha":str(tempalpha),
"l1weight":str(l1weight),
"gctb_ld_model":panel,
"Train_pure_prs":roc_auc_score(phenotype_train["Phenotype"].values,prs_train['SCORE'].values),
"Train_null_model":roc_auc_score(phenotype_train["Phenotype"].values,train_null_predicted.values),
"Train_best_model":roc_auc_score(phenotype_train["Phenotype"].values,train_best_predicted.values),
"Test_pure_prs":roc_auc_score(phenotype_test["Phenotype"].values,prs_test['SCORE'].values),
"Test_null_model":roc_auc_score(phenotype_test["Phenotype"].values,test_null_predicted.values),
"Test_best_model":roc_auc_score(phenotype_test["Phenotype"].values,test_best_predicted.values),
}, ignore_index=True)
prs_result.to_csv(traindirec+os.sep+Name+os.sep+"Results.csv",index=False)
return
# This function fit the binary model on the PRS.
def fit_continous_phenotype_on_PRS(traindirec, newtrainfilename,panel, p,p1_val, p2_val, p3_val, c1_val, c2_val, c3_val,Name,pvaluefile):
threshold_values = allpvalues
# Merge the covariates, pca and phenotypes.
tempphenotype_train = pd.read_table(traindirec+os.sep+newtrainfilename+".clumped.pruned"+".fam", sep="\s+",header=None)
phenotype_train = pd.DataFrame()
phenotype_train["Phenotype"] = tempphenotype_train[5].values
pcs_train = pd.read_table(traindirec+os.sep+trainfilename+".eigenvec", sep="\s+",header=None, names=["FID", "IID"] + [f"PC{str(i)}" for i in range(1, int(p)+1)])
covariate_train = pd.read_table(traindirec+os.sep+trainfilename+".cov",sep="\s+")
covariate_train.fillna(0, inplace=True)
covariate_train = covariate_train[covariate_train["FID"].isin(pcs_train["FID"].values) & covariate_train["IID"].isin(pcs_train["IID"].values)]
covariate_train['FID'] = covariate_train['FID'].astype(str)
pcs_train['FID'] = pcs_train['FID'].astype(str)
covariate_train['IID'] = covariate_train['IID'].astype(str)
pcs_train['IID'] = pcs_train['IID'].astype(str)
covandpcs_train = pd.merge(covariate_train, pcs_train, on=["FID","IID"])
covandpcs_train.fillna(0, inplace=True)
## Scale the covariates!
from sklearn.preprocessing import MinMaxScaler
from sklearn.metrics import explained_variance_score
scaler = MinMaxScaler()
normalized_values_train = scaler.fit_transform(covandpcs_train.iloc[:, 2:])
#covandpcs_train.iloc[:, 2:] = normalized_values_test
tempphenotype_test = pd.read_table(traindirec+os.sep+testfilename+".clumped.pruned"+".fam", sep="\s+",header=None)
phenotype_test= pd.DataFrame()
phenotype_test["Phenotype"] = tempphenotype_test[5].values
pcs_test = pd.read_table(traindirec+os.sep+testfilename+".eigenvec", sep="\s+",header=None, names=["FID", "IID"] + [f"PC{str(i)}" for i in range(1, int(p)+1)])
covariate_test = pd.read_table(traindirec+os.sep+testfilename+".cov",sep="\s+")
covariate_test.fillna(0, inplace=True)
covariate_test = covariate_test[covariate_test["FID"].isin(pcs_test["FID"].values) & covariate_test["IID"].isin(pcs_test["IID"].values)]
covariate_test['FID'] = covariate_test['FID'].astype(str)
pcs_test['FID'] = pcs_test['FID'].astype(str)
covariate_test['IID'] = covariate_test['IID'].astype(str)
pcs_test['IID'] = pcs_test['IID'].astype(str)
covandpcs_test = pd.merge(covariate_test, pcs_test, on=["FID","IID"])
covandpcs_test.fillna(0, inplace=True)
normalized_values_test = scaler.transform(covandpcs_test.iloc[:, 2:])
#covandpcs_test.iloc[:, 2:] = normalized_values_test
tempalphas = [0.1,0.2,0.3,0.4,0.5,0.6,0.7,0.8,0.9]
l1weights = [0.1,0.2,0.3,0.4,0.5,0.6,0.7,0.8,0.9]
tempalphas = [0.1]
l1weights = [0.1]
#phenotype_train["Phenotype"] = phenotype_train["Phenotype"].replace({1: 0, 2: 1})
#phenotype_test["Phenotype"] = phenotype_test["Phenotype"].replace({1: 0, 2: 1})
for tempalpha in tempalphas:
for l1weight in l1weights:
try:
#null_model = sm.OLS(phenotype_train["Phenotype"], sm.add_constant(covandpcs_train.iloc[:, 2:])).fit_regularized(alpha=tempalpha, L1_wt=l1weight)
null_model = sm.OLS(phenotype_train["Phenotype"], sm.add_constant(covandpcs_train.iloc[:, 2:])).fit()
#null_model = sm.OLS(phenotype_train["Phenotype"], sm.add_constant(covandpcs_train.iloc[:, 2:])).fit()
except:
print("XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX")
continue
train_null_predicted = null_model.predict(sm.add_constant(covandpcs_train.iloc[:, 2:]))
from sklearn.metrics import roc_auc_score, confusion_matrix
from sklearn.metrics import r2_score
test_null_predicted = null_model.predict(sm.add_constant(covandpcs_test.iloc[:, 2:]))
global prs_result
for i in threshold_values:
try:
prs_train = pd.read_table(traindirec+os.sep+Name+os.sep+"train_data.pv_"+f"{i}.profile", sep="\s+", usecols=["FID", "IID", "SCORE"])
except:
continue
prs_train['FID'] = prs_train['FID'].astype(str)
prs_train['IID'] = prs_train['IID'].astype(str)
try:
prs_test = pd.read_table(traindirec+os.sep+Name+os.sep+"test_data.pv_"+f"{i}.profile", sep="\s+", usecols=["FID", "IID", "SCORE"])
except:
continue
prs_test['FID'] = prs_test['FID'].astype(str)
prs_test['IID'] = prs_test['IID'].astype(str)
pheno_prs_train = pd.merge(covandpcs_train, prs_train, on=["FID", "IID"])
pheno_prs_test = pd.merge(covandpcs_test, prs_test, on=["FID", "IID"])
try:
#model = sm.OLS(phenotype_train["Phenotype"], sm.add_constant(pheno_prs_train.iloc[:, 2:])).fit_regularized(alpha=tempalpha, L1_wt=l1weight)
model = sm.OLS(phenotype_train["Phenotype"], sm.add_constant(pheno_prs_train.iloc[:, 2:])).fit()
except:
continue
train_best_predicted = model.predict(sm.add_constant(pheno_prs_train.iloc[:, 2:]))
test_best_predicted = model.predict(sm.add_constant(pheno_prs_test.iloc[:, 2:]))
from sklearn.metrics import roc_auc_score, confusion_matrix
prs_result = prs_result._append({
"clump_p1": c1_val,
"clump_r2": c2_val,
"clump_kb": c3_val,
"p_window_size": p1_val,
"p_slide_size": p2_val,
"p_LD_threshold": p3_val,
"pvalue": i,
"numberofpca":p,
"tempalpha":str(tempalpha),
"l1weight":str(l1weight),
"gctb_ld_model":panel,
"Train_pure_prs":explained_variance_score(phenotype_train["Phenotype"],prs_train['SCORE'].values),
"Train_null_model":explained_variance_score(phenotype_train["Phenotype"],train_null_predicted),
"Train_best_model":explained_variance_score(phenotype_train["Phenotype"],train_best_predicted),
"Test_pure_prs":explained_variance_score(phenotype_test["Phenotype"],prs_test['SCORE'].values),
"Test_null_model":explained_variance_score(phenotype_test["Phenotype"],test_null_predicted),
"Test_best_model":explained_variance_score(phenotype_test["Phenotype"],test_best_predicted),
}, ignore_index=True)
prs_result.to_csv(traindirec+os.sep+Name+os.sep+"Results.csv",index=False)
return
Execute GCTB-SBayesRC#
def transform_SBayesRC_data(traindirec, newtrainfilename,panel,p, p1_val, p2_val, p3_val, c1_val, c2_val, c3_val,Name,pvaluefile):
### First perform clumping on the file and save the clumpled file.
#perform_clumping_and_pruning_on_individual_data(traindirec, newtrainfilename,p, p1_val, p2_val, p3_val, c1_val, c2_val, c3_val,Name,pvaluefile)
# Also extract the PCA at this point for both test and training data.
#calculate_pca_for_traindata_testdata_for_clumped_pruned_snps(traindirec, newtrainfilename,p)
#Extract p-values from the GWAS file.
os.system("awk "+"\'"+"{print $3,$8}"+"\'"+" ./"+filedirec+os.sep+filedirec+".txt > ./"+traindirec+os.sep+"SNP.pvalue")
# At this stage, we will merge the PCA and COV file.
tempphenotype_train = pd.read_table(traindirec+os.sep+newtrainfilename+".clumped.pruned"+".fam", sep="\s+",header=None)
phenotype = pd.DataFrame()
phenotype = tempphenotype_train[[0,1,5]]
phenotype.to_csv(traindirec+os.sep+trainfilename+".PHENO",sep="\t",header=['FID', 'IID', 'PHENO'],index=False)
pcs_train = pd.read_table(traindirec+os.sep+trainfilename+".eigenvec", sep="\s+",header=None, names=["FID", "IID"] + [f"PC{str(i)}" for i in range(1, int(p)+1)])
covariate_train = pd.read_table(traindirec+os.sep+trainfilename+".cov",sep="\s+")
covariate_train.fillna(0, inplace=True)
print(covariate_train.head())
print(len(covariate_train))
covariate_train = covariate_train[covariate_train["FID"].isin(pcs_train["FID"].values) & covariate_train["IID"].isin(pcs_train["IID"].values)]
print(len(covariate_train))
covariate_train['FID'] = covariate_train['FID'].astype(str)
pcs_train['FID'] = pcs_train['FID'].astype(str)
covariate_train['IID'] = covariate_train['IID'].astype(str)
pcs_train['IID'] = pcs_train['IID'].astype(str)
covandpcs_train = pd.merge(covariate_train, pcs_train, on=["FID","IID"])
covandpcs_train.to_csv(traindirec+os.sep+trainfilename+".COV_PCA",sep="\t",index=False)
# Define the paths to the files
# Delete the files generated in the previous iterations.
files_to_remove = [
traindirec+os.sep+"SBayesRC.snpRes"
]
# Loop through the files and remove them if they exist
for file_path in files_to_remove:
if os.path.exists(file_path):
os.remove(file_path)
print(f"Removed: {file_path}")
else:
print(f"File does not exist: {file_path}")
for chromosome in range(1, 23):
# Plink command to split by chromosome
plink_command = [
"./plink",
"--bfile", traindirec+os.sep+newtrainfilename+".clumped.pruned",
"--chr", str(chromosome),
"--make-bed",
"--out", traindirec+os.sep+newtrainfilename+".clumped.pruned."+str(chromosome),
]
# Run the Plink command
#subprocess.run(plink_command, check=True)
ne = pd.read_csv(traindirec+os.sep+newtrainfilename+".clumped.pruned.fam")
ne = len(ne)
ldpath = "SBayesR_LDFrames/"
#Step 1: QC & imputation of summary statistics for SNPs in LD reference but not in GWAS data.
#gctb --ldm-eigen ldm --gwas-summary test.ma --impute-summary --out test --thread 4
# This will increase the number of SNPs in the orginial GWAS file and betas for the GWAS files are imputed.
#
dir_name = traindirec+os.sep+"LDfull"
# Check if the directory exists, if not, create it
if not os.path.exists(dir_name):
os.mkdir(dir_name)
print(f"Directory '{dir_name}' created successfully.")
else:
print(f"Directory '{dir_name}' already exists.")
command = [
"./gctb",
"--ldm-eigen", ldpath+os.sep+panel, # Specify the Panel path.
"--gwas-summary", filedirec+os.sep+"SBayesR.ma",
"--impute-summary",
"--out", traindirec+os.sep+"LDfull"+os.sep+"test"
]
# Run the command
subprocess.run(command)
#raise
#Step 2: Run SBayesRC with annotation data.
#gctb --ldm-eigen ldm --gwas-summary test.imputed.ma --sbayes RC --annot annot.txt --out test --thread 4
command = [
"./gctb",
"--ldm-eigen", ldpath+os.sep+panel,
"--gwas-summary", traindirec+os.sep+"LDfull"+os.sep+"test.imputed.ma",
#"--gwas-summary", filedirec+os.sep+"SBayesR.ma",
"--sbayes", "RC",
"--annot", ldpath+"annot_baseline2.2.txt",
"--out", traindirec+os.sep+"SBayesRC"
]
subprocess.run(command)
allgwasdirec = traindirec+os.sep+"SBayesRC.snpRes"
try:
tempgwas = pd.read_csv(allgwasdirec,sep="\s+" )
except:
print("GWAS not generated!")
return
if check_phenotype_is_binary_or_continous(filedirec)=="Binary":
tempgwas["A1Effect"] = np.exp(tempgwas["A1Effect"])
tempgwas = tempgwas.fillna(0)
tempgwas = tempgwas.replace([np.inf, -np.inf], 0)
else:
pass
# Select columns 2, 5, and 8 (Python uses zero-based indexing, so adjust accordingly)
selected_columns = tempgwas.iloc[:, [1, 4, 7]]
# Save the selected columns to a file
selected_columns.to_csv(allgwasdirec+"_modified", sep="\t", index=False)
command = [
"./plink",
"--bfile", traindirec+os.sep+newtrainfilename+".clumped.pruned",
### SNP column = 1, Effect allele column 2 = 4, Effect column=4
"--score", allgwasdirec+"_modified", "1", "2", "3", "header",
"--q-score-range", traindirec+os.sep+"range_list",traindirec+os.sep+"SNP.pvalue",
#"--extract", traindirec+os.sep+trainfilename+".valid.snp",
"--out", traindirec+os.sep+Name+os.sep+trainfilename
]
#exit(0)
subprocess.run(command)
command = [
"./plink",
"--bfile", folddirec+os.sep+testfilename,
### SNP column = 3, Effect allele column 1 = 4, Beta column=12
"--score", allgwasdirec+"_modified", "1", "2", "3", "header",
"--q-score-range", traindirec+os.sep+"range_list",traindirec+os.sep+"SNP.pvalue",
"--out", folddirec+os.sep+Name+os.sep+testfilename
]
subprocess.run(command)
if check_phenotype_is_binary_or_continous(filedirec)=="Binary":
print("Binary Phenotype!")
fit_binary_phenotype_on_PRS(traindirec, newtrainfilename,panel, p, p1_val, p2_val, p3_val, c1_val, c2_val, c3_val,Name,pvaluefile)
else:
print("Continous Phenotype!")
fit_continous_phenotype_on_PRS(traindirec, newtrainfilename,panel, p,p1_val, p2_val, p3_val, c1_val, c2_val, c3_val,Name,pvaluefile)
return
ldpanels = ["ukbEUR_HM3","ukbEUR_Imputed"]
ldpanels = ["ukbEUR_HM3"]
result_directory = "GCTB-SBayesRC"
# Nested loops to iterate over different parameter values
create_directory(folddirec+os.sep+result_directory)
for p1_val in p_window_size:
for p2_val in p_slide_size:
for p3_val in p_LD_threshold:
for c1_val in clump_p1:
for c2_val in clump_r2:
for c3_val in clump_kb:
for p in numberofpca:
for panel in ldpanels:
#for bayesmodel in bayesmodels:
transform_SBayesRC_data(folddirec, newtrainfilename,panel, p, str(p1_val), str(p2_val), str(p3_val), str(c1_val), str(c2_val), str(c3_val), result_directory, pvaluefile)
FID IID Sex
0 HG00097 HG00097 2
1 HG00099 HG00099 2
2 HG00101 HG00101 1
3 HG00102 HG00102 2
4 HG00103 HG00103 1
380
380
File does not exist: SampleData1/Fold_0/SBayesRC.snpRes
Directory 'SampleData1/Fold_0/LDfull' already exists.
******************************************************************
* GCTB 2.05beta *
* Genome-wide Complex Trait Bayesian analysis *
* Authors: Jian Zeng, Luke Lloyd-Jones, Zhili Zheng, Shouye Liu *
* MIT License *
******************************************************************
Analysis started: Sat Sep 7 17:06:16 2024
Options:
--ldm-eigen SBayesR_LDFrames//ukbEUR_HM3
--gwas-summary SampleData1/SBayesR.ma
--impute-summary
--out SampleData1/Fold_0/LDfull/test
Reading LD matrix eigen-decomposition data...
Reading LDM info from file [SBayesR_LDFrames//ukbEUR_HM3/ldm.info].
591 LD Blocks to be included from [SBayesR_LDFrames//ukbEUR_HM3/ldm.info].
Reading LDM SNP info from file [SBayesR_LDFrames//ukbEUR_HM3/snp.info].
1154522 SNPs to be included from [SBayesR_LDFrames//ukbEUR_HM3/snp.info].
Reading GWAS summary data from [SampleData1/SBayesR.ma].
flipped 38414 SNPs according to the minor allele in the reference and GWAS samples.
137219 matched SNPs in the GWAS summary data (in total 499617 SNPs).
Imputing summary statistics for 1017303 SNPs in the LD reference but not in the GWAS data file...
Imputation of summary statistics is completed (time used: 0:2:43).
Summary statistics of all SNPs are save into file [SampleData1/Fold_0/LDfull/test.imputed.ma].
Analysis finished: Sat Sep 7 17:09:05 2024
Computational time: 0:2:49
******************************************************************
* GCTB 2.05beta *
* Genome-wide Complex Trait Bayesian analysis *
* Authors: Jian Zeng, Luke Lloyd-Jones, Zhili Zheng, Shouye Liu *
* MIT License *
******************************************************************
Analysis started: Sat Sep 7 17:09:05 2024
Options:
--ldm-eigen SBayesR_LDFrames//ukbEUR_HM3
--gwas-summary SampleData1/Fold_0/LDfull/test.imputed.ma
--sbayes RC
--annot SBayesR_LDFrames/annot_baseline2.2.txt
--out SampleData1/Fold_0/SBayesRC
Reading LD matrix eigen-decomposition data...
Reading LDM info from file [SBayesR_LDFrames//ukbEUR_HM3/ldm.info].
591 LD Blocks to be included from [SBayesR_LDFrames//ukbEUR_HM3/ldm.info].
Reading LDM SNP info from file [SBayesR_LDFrames//ukbEUR_HM3/snp.info].
1154522 SNPs to be included from [SBayesR_LDFrames//ukbEUR_HM3/snp.info].
Reading SNP annotation from [SBayesR_LDFrames/annot_baseline2.2.txt].
1154522 matched SNPs in the annotation file (97 annotations and 1154522 SNPs have more than one annotation).
Reading GWAS summary data from [SampleData1/Fold_0/LDfull/test.imputed.ma].
1154522 matched SNPs in the GWAS summary data (in total 1154522 SNPs).
1154522 SNPs on 22 chromosomes are included.
591 LD blocks are included.
Data summary:
mean sd
GWAS SNP Phenotypic variance 1.153 0.164
GWAS SNP heterozygosity 0.325 0.145
GWAS SNP sample size 388028 0
GWAS SNP effect (in genotype SD unit) 0.000 0.003
GWAS SNP SE 0.002 0.000
LD block size 1953.506 656.095
LD block rank 696.164 166.350
Finding the best eigen cutoff from [0.995 0.99 0.95 0.9] based on pseudo summary data validation.
Cutoff Prediction accuracy (r) Relative accuracy
0.995 1.13879 1
0.99 1.11997 0.983476
0.95 1.01985 0.895553
0.9 0.852343 0.748464
0.995 is selected to be the eigen cutoff to continue the analysis (time used: 0:5:55).
SBayesRC
Using the low-rank model
Gamma: 0 0.001 0.01 0.1 1
Fitting model assuming scaled genotypes
MCMC launched ...
Chain length: 3000 iterations
Burn-in: 1000 iterations
Iter NumSnp1 NumSnp2 NumSnp3 NumSnp4 NumSnp5 SigmaSq ResVar GenVar hsq Rounding TimeLeft
100 1022769 130202 1528 23 0 0.0070 0.0905 0.7308 0.6211 0.0000 2:22:6
200 1016611 136623 1265 23 0 0.0072 0.0826 0.7391 0.6281 0.0000 2:21:52
300 1015018 138235 1236 33 0 0.0072 0.0802 0.7409 0.6296 0.0000 2:18:18
400 1015067 137973 1443 39 0 0.0073 0.0798 0.7420 0.6306 0.0000 2:14:0
500 1015201 137885 1386 50 0 0.0073 0.0790 0.7446 0.6328 0.0000 2:9:15
600 1014533 138501 1449 39 0 0.0073 0.0777 0.7422 0.6308 0.0000 2:4:20
700 1014430 138738 1308 46 0 0.0073 0.0777 0.7423 0.6308 0.0000 1:59:6
800 1014631 138680 1168 43 0 0.0073 0.0777 0.7420 0.6306 0.0000 1:53:45
900 1014978 138065 1426 53 0 0.0073 0.0775 0.7431 0.6315 0.0000 1:48:27
1000 1014395 138817 1269 41 0 0.0073 0.0766 0.7437 0.6320 0.0000 1:43:8
1100 1013673 139483 1323 41 2 0.0073 0.0777 0.7435 0.6319 0.0000 1:37:51
1200 1014609 138532 1311 70 0 0.0073 0.0758 0.7437 0.6320 0.0000 1:32:33
1300 1014725 138467 1280 50 0 0.0072 0.0756 0.7417 0.6303 0.0000 1:27:17
1400 1015175 137835 1469 43 0 0.0073 0.0756 0.7445 0.6327 0.0000 1:22:1
1500 1015133 137962 1383 44 0 0.0073 0.0757 0.7419 0.6305 0.0000 1:16:49
1600 1014213 138842 1433 34 0 0.0073 0.0751 0.7428 0.6313 0.0000 1:11:36
1700 1014805 138312 1374 31 0 0.0073 0.0761 0.7422 0.6308 0.0000 1:6:24
1800 1014891 138273 1318 40 0 0.0073 0.0749 0.7427 0.6312 0.0000 1:1:17
1900 1014627 138536 1328 31 0 0.0073 0.0758 0.7428 0.6312 0.0000 0:56:8
2000 1014375 138787 1329 31 0 0.0073 0.0755 0.7421 0.6307 0.0000 0:51:1
2100 1014632 138697 1157 36 0 0.0073 0.0776 0.7414 0.6301 0.0000 0:45:54
2200 1014079 139034 1370 39 0 0.0073 0.0767 0.7432 0.6316 0.0000 0:40:48
2300 1014532 138634 1322 34 0 0.0073 0.0772 0.7417 0.6304 0.0000 0:35:41
2400 1015466 137793 1222 41 0 0.0073 0.0775 0.7426 0.6311 0.0000 0:30:34
2500 1015116 138086 1282 38 0 0.0073 0.0776 0.7442 0.6324 0.0000 0:25:28
2600 1015229 137987 1267 39 0 0.0074 0.0776 0.7431 0.6316 0.0000 0:20:22
2700 1013925 139217 1321 59 0 0.0073 0.0776 0.7421 0.6307 0.0000 0:15:16
2800 1014825 138385 1269 43 0 0.0073 0.0771 0.7428 0.6312 0.0000 0:10:10
2900 1014059 139229 1197 37 0 0.0073 0.0776 0.7432 0.6316 0.0000 0:5:5
3000 1013998 139173 1303 48 0 0.0073 0.0769 0.7441 0.6323 0.0000 0:0:0
MCMC cycles completed.
Posterior statistics from MCMC samples:
Mean SD
NumSnp1 1014631.687500 461.146973
NumSnp2 138543.687500 487.961334
NumSnp3 1306.479980 79.679039
NumSnp4 40.299999 7.807688
NumSnp5 0.005000 0.070534
SigmaSq 0.007285 0.000035
ResVar 0.076645 0.001160
GenVar 0.742690 0.000834
hsq 0.631173 0.000709
Analysis finished: Sat Sep 7 19:49:03 2024
Computational time: 2:39:58
PLINK v1.90b7.2 64-bit (11 Dec 2023) www.cog-genomics.org/plink/1.9/
(C) 2005-2023 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to SampleData1/Fold_0/GCTB-SBayesRC/train_data.log.
Options in effect:
--bfile SampleData1/Fold_0/train_data.QC.clumped.pruned
--out SampleData1/Fold_0/GCTB-SBayesRC/train_data
--q-score-range SampleData1/Fold_0/range_list SampleData1/Fold_0/SNP.pvalue
--score SampleData1/Fold_0/SBayesRC.snpRes_modified 1 2 3 header
63761 MB RAM detected; reserving 31880 MB for main workspace.
172878 variants loaded from .bim file.
380 people (183 males, 197 females) loaded from .fam.
380 phenotype values loaded from .fam.
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 380 founders and 0 nonfounders present.
Calculating allele frequencies... 10111213141516171819202122232425262728293031323334353637383940414243444546474849505152535455565758596061626364656667686970717273747576777879808182838485868788899091929394959697989 done.
Total genotyping rate is 0.999891.
172878 variants and 380 people pass filters and QC.
Phenotype data is quantitative.
--score: 37801 valid predictors loaded.
Warning: 1116721 lines skipped in --score file (1116721 due to variant ID
mismatch, 0 due to allele code mismatch); see
SampleData1/Fold_0/GCTB-SBayesRC/train_data.nopred for details.
Warning: 461817 lines skipped in --q-score-range data file.
--score: 20 ranges processed.
Results written to SampleData1/Fold_0/GCTB-SBayesRC/train_data.*.profile.
PLINK v1.90b7.2 64-bit (11 Dec 2023) www.cog-genomics.org/plink/1.9/
(C) 2005-2023 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to SampleData1/Fold_0/GCTB-SBayesRC/test_data.log.
Options in effect:
--bfile SampleData1/Fold_0/test_data
--out SampleData1/Fold_0/GCTB-SBayesRC/test_data
--q-score-range SampleData1/Fold_0/range_list SampleData1/Fold_0/SNP.pvalue
--score SampleData1/Fold_0/SBayesRC.snpRes_modified 1 2 3 header
63761 MB RAM detected; reserving 31880 MB for main workspace.
551892 variants loaded from .bim file.
95 people (44 males, 51 females) loaded from .fam.
95 phenotype values loaded from .fam.
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 95 founders and 0 nonfounders present.
Calculating allele frequencies... 10111213141516171819202122232425262728293031323334353637383940414243444546474849505152535455565758596061626364656667686970717273747576777879808182838485868788899091929394959697989 done.
Total genotyping rate is 0.999896.
551892 variants and 95 people pass filters and QC.
Phenotype data is quantitative.
Warning: 1017306 lines skipped in --score file (1017303 due to variant ID
mismatch, 3 due to allele code mismatch); see
SampleData1/Fold_0/GCTB-SBayesRC/test_data.nopred for details.
--score: 137216 valid predictors loaded.
Warning: 362402 lines skipped in --q-score-range data file.
--score: 20 ranges processed.
Results written to SampleData1/Fold_0/GCTB-SBayesRC/test_data.*.profile.
Continous Phenotype!
/tmp/ipykernel_1232748/672316298.py:348: FutureWarning: The behavior of DataFrame concatenation with empty or all-NA entries is deprecated. In a future version, this will no longer exclude empty or all-NA columns when determining the result dtypes. To retain the old behavior, exclude the relevant entries before the concat operation.
prs_result = prs_result._append({
Repeat the process for each fold.#
Change the foldnumber variable.
#foldnumber = sys.argv[1]
foldnumber = "0" # Setting 'foldnumber' to "0"
Or uncomment the following line:
# foldnumber = sys.argv[1]
python GCTB-SBayesRC.py 0
python GCTB-SBayesRC.py 1
python GCTB-SBayesRC.py 2
python GCTB-SBayesRC.py 3
python GCTB-SBayesRC.py 4
The following files should exist after the execution:
SampleData1/Fold_0/GCTB-SBayesRC/Results.csvSampleData1/Fold_1/GCTB-SBayesRC/Results.csvSampleData1/Fold_2/GCTB-SBayesRC/Results.csvSampleData1/Fold_3/GCTB-SBayesRC/Results.csvSampleData1/Fold_4/GCTB-SBayesRC/Results.csv
Check the results file for each fold.#
import os
result_directory = "GCTB-SBayesRC"
# List of file names to check for existence
f = [
"./"+filedirec+"/Fold_0"+os.sep+result_directory+"Results.csv",
"./"+filedirec+"/Fold_1"+os.sep+result_directory+"Results.csv",
"./"+filedirec+"/Fold_2"+os.sep+result_directory+"Results.csv",
"./"+filedirec+"/Fold_3"+os.sep+result_directory+"Results.csv",
"./"+filedirec+"/Fold_4"+os.sep+result_directory+"Results.csv",
]
# Loop through each file name in the list
for loop in range(0,5):
# Check if the file exists in the specified directory for the given fold
if os.path.exists(filedirec+os.sep+"Fold_"+str(loop)+os.sep+result_directory+os.sep+"Results.csv"):
temp = pd.read_csv(filedirec+os.sep+"Fold_"+str(loop)+os.sep+result_directory+os.sep+"Results.csv")
print("Fold_",loop, "Yes, the file exists.")
#print(temp.head())
print("Number of P-values processed: ",len(temp))
# Print a message indicating that the file exists
else:
# Print a message indicating that the file does not exist
print("Fold_",loop, "No, the file does not exist.")
Fold_ 0 Yes, the file exists.
Number of P-values processed: 20
Fold_ 1 Yes, the file exists.
Number of P-values processed: 20
Fold_ 2 Yes, the file exists.
Number of P-values processed: 20
Fold_ 3 Yes, the file exists.
Number of P-values processed: 20
Fold_ 4 Yes, the file exists.
Number of P-values processed: 20
Sum the results for each fold.#
print("We have to ensure when we sum the entries across all Folds, the same rows are merged!")
def sum_and_average_columns(data_frames):
"""Sum and average numerical columns across multiple DataFrames, and keep non-numerical columns unchanged."""
# Initialize DataFrame to store the summed results for numerical columns
summed_df = pd.DataFrame()
non_numerical_df = pd.DataFrame()
for df in data_frames:
# Identify numerical and non-numerical columns
numerical_cols = df.select_dtypes(include=[np.number]).columns
non_numerical_cols = df.select_dtypes(exclude=[np.number]).columns
# Sum numerical columns
if summed_df.empty:
summed_df = pd.DataFrame(0, index=range(len(df)), columns=numerical_cols)
summed_df[numerical_cols] = summed_df[numerical_cols].add(df[numerical_cols], fill_value=0)
# Keep non-numerical columns (take the first non-numerical entry for each column)
if non_numerical_df.empty:
non_numerical_df = df[non_numerical_cols]
else:
non_numerical_df[non_numerical_cols] = non_numerical_df[non_numerical_cols].combine_first(df[non_numerical_cols])
# Divide the summed values by the number of dataframes to get the average
averaged_df = summed_df / len(data_frames)
# Combine numerical and non-numerical DataFrames
result_df = pd.concat([averaged_df, non_numerical_df], axis=1)
return result_df
from functools import reduce
import os
import pandas as pd
from functools import reduce
def find_common_rows(allfoldsframe):
# Define the performance columns that need to be excluded
performance_columns = [
'Train_null_model', 'Train_pure_prs', 'Train_best_model',
'Test_pure_prs', 'Test_null_model', 'Test_best_model'
]
important_columns = [
'clump_p1',
'clump_r2',
'clump_kb',
'p_window_size',
'p_slide_size',
'p_LD_threshold',
'pvalue',
#'h2model',
'gctb_ld_model',
'BayesModel',
'numberofpca',
'tempalpha',
'l1weight',
]
# Function to remove performance columns from a DataFrame
def drop_performance_columns(df):
return df.drop(columns=performance_columns, errors='ignore')
def get_important_columns(df ):
existing_columns = [col for col in important_columns if col in df.columns]
if existing_columns:
return df[existing_columns].copy()
else:
return pd.DataFrame()
# Drop performance columns from all DataFrames in the list
allfoldsframe_dropped = [drop_performance_columns(df) for df in allfoldsframe]
# Get the important columns.
allfoldsframe_dropped = [get_important_columns(df) for df in allfoldsframe_dropped]
# Iteratively find common rows and track unique and common rows
common_rows = allfoldsframe_dropped[0]
print(common_rows.head())
for i in range(1, len(allfoldsframe_dropped)):
# Get the next DataFrame
next_df = allfoldsframe_dropped[i]
# Count unique rows in the current DataFrame and the next DataFrame
unique_in_common = common_rows.shape[0]
unique_in_next = next_df.shape[0]
# Find common rows between the current common_rows and the next DataFrame
common_rows = pd.merge(common_rows, next_df, how='inner')
print(common_rows.head())
# Count the common rows after merging
common_count = common_rows.shape[0]
# Print the unique and common row counts
print(f"Iteration {i}:")
print(f"Unique rows in current common DataFrame: {unique_in_common}")
print(f"Unique rows in next DataFrame: {unique_in_next}")
print(f"Common rows after merge: {common_count}\n")
# Now that we have the common rows, extract these from the original DataFrames
extracted_common_rows_frames = []
for original_df in allfoldsframe:
# Merge the common rows with the original DataFrame, keeping only the rows that match the common rows
extracted_common_rows = pd.merge(common_rows, original_df, how='inner', on=common_rows.columns.tolist())
# Add the DataFrame with the extracted common rows to the list
extracted_common_rows_frames.append(extracted_common_rows)
# Print the number of rows in the common DataFrames
for i, df in enumerate(extracted_common_rows_frames):
print(f"DataFrame {i + 1} with extracted common rows has {df.shape[0]} rows.")
# Return the list of DataFrames with extracted common rows
return extracted_common_rows_frames
# Example usage (assuming allfoldsframe is populated as shown earlier):
allfoldsframe = []
# Loop through each file name in the list
for loop in range(0, 5):
# Check if the file exists in the specified directory for the given fold
file_path = os.path.join(filedirec, "Fold_" + str(loop), result_directory, "Results.csv")
if os.path.exists(file_path):
allfoldsframe.append(pd.read_csv(file_path))
# Print a message indicating that the file exists
print("Fold_", loop, "Yes, the file exists.")
else:
# Print a message indicating that the file does not exist
print("Fold_", loop, "No, the file does not exist.")
# Find the common rows across all folds and return the list of extracted common rows
extracted_common_rows_list = find_common_rows(allfoldsframe)
# Sum the values column-wise
# For string values, do not sum it the values are going to be the same for each fold.
# Only sum the numeric values.
divided_result = sum_and_average_columns(extracted_common_rows_list)
print(divided_result)
We have to ensure when we sum the entries across all Folds, the same rows are merged!
Fold_ 0 Yes, the file exists.
Fold_ 1 Yes, the file exists.
Fold_ 2 Yes, the file exists.
Fold_ 3 Yes, the file exists.
Fold_ 4 Yes, the file exists.
clump_p1 clump_r2 clump_kb p_window_size p_slide_size p_LD_threshold \
0 1 0.1 200 200 50 0.25
1 1 0.1 200 200 50 0.25
2 1 0.1 200 200 50 0.25
3 1 0.1 200 200 50 0.25
4 1 0.1 200 200 50 0.25
pvalue gctb_ld_model numberofpca tempalpha l1weight
0 1.000000e-10 ukbEUR_HM3 6 0.1 0.1
1 3.359818e-10 ukbEUR_HM3 6 0.1 0.1
2 1.128838e-09 ukbEUR_HM3 6 0.1 0.1
3 3.792690e-09 ukbEUR_HM3 6 0.1 0.1
4 1.274275e-08 ukbEUR_HM3 6 0.1 0.1
clump_p1 clump_r2 clump_kb p_window_size p_slide_size p_LD_threshold \
0 1 0.1 200 200 50 0.25
1 1 0.1 200 200 50 0.25
2 1 0.1 200 200 50 0.25
3 1 0.1 200 200 50 0.25
4 1 0.1 200 200 50 0.25
pvalue gctb_ld_model numberofpca tempalpha l1weight
0 1.000000e-10 ukbEUR_HM3 6 0.1 0.1
1 3.359818e-10 ukbEUR_HM3 6 0.1 0.1
2 1.128838e-09 ukbEUR_HM3 6 0.1 0.1
3 3.792690e-09 ukbEUR_HM3 6 0.1 0.1
4 1.274275e-08 ukbEUR_HM3 6 0.1 0.1
Iteration 1:
Unique rows in current common DataFrame: 20
Unique rows in next DataFrame: 20
Common rows after merge: 20
clump_p1 clump_r2 clump_kb p_window_size p_slide_size p_LD_threshold \
0 1 0.1 200 200 50 0.25
1 1 0.1 200 200 50 0.25
2 1 0.1 200 200 50 0.25
3 1 0.1 200 200 50 0.25
4 1 0.1 200 200 50 0.25
pvalue gctb_ld_model numberofpca tempalpha l1weight
0 1.000000e-10 ukbEUR_HM3 6 0.1 0.1
1 3.359818e-10 ukbEUR_HM3 6 0.1 0.1
2 1.128838e-09 ukbEUR_HM3 6 0.1 0.1
3 3.792690e-09 ukbEUR_HM3 6 0.1 0.1
4 1.274275e-08 ukbEUR_HM3 6 0.1 0.1
Iteration 2:
Unique rows in current common DataFrame: 20
Unique rows in next DataFrame: 20
Common rows after merge: 20
clump_p1 clump_r2 clump_kb p_window_size p_slide_size p_LD_threshold \
0 1 0.1 200 200 50 0.25
1 1 0.1 200 200 50 0.25
2 1 0.1 200 200 50 0.25
3 1 0.1 200 200 50 0.25
4 1 0.1 200 200 50 0.25
pvalue gctb_ld_model numberofpca tempalpha l1weight
0 1.000000e-10 ukbEUR_HM3 6 0.1 0.1
1 3.359818e-10 ukbEUR_HM3 6 0.1 0.1
2 1.128838e-09 ukbEUR_HM3 6 0.1 0.1
3 3.792690e-09 ukbEUR_HM3 6 0.1 0.1
4 1.274275e-08 ukbEUR_HM3 6 0.1 0.1
Iteration 3:
Unique rows in current common DataFrame: 20
Unique rows in next DataFrame: 20
Common rows after merge: 20
clump_p1 clump_r2 clump_kb p_window_size p_slide_size p_LD_threshold \
0 1 0.1 200 200 50 0.25
1 1 0.1 200 200 50 0.25
2 1 0.1 200 200 50 0.25
3 1 0.1 200 200 50 0.25
4 1 0.1 200 200 50 0.25
pvalue gctb_ld_model numberofpca tempalpha l1weight
0 1.000000e-10 ukbEUR_HM3 6 0.1 0.1
1 3.359818e-10 ukbEUR_HM3 6 0.1 0.1
2 1.128838e-09 ukbEUR_HM3 6 0.1 0.1
3 3.792690e-09 ukbEUR_HM3 6 0.1 0.1
4 1.274275e-08 ukbEUR_HM3 6 0.1 0.1
Iteration 4:
Unique rows in current common DataFrame: 20
Unique rows in next DataFrame: 20
Common rows after merge: 20
DataFrame 1 with extracted common rows has 20 rows.
DataFrame 2 with extracted common rows has 20 rows.
DataFrame 3 with extracted common rows has 20 rows.
DataFrame 4 with extracted common rows has 20 rows.
DataFrame 5 with extracted common rows has 20 rows.
clump_p1 clump_r2 clump_kb p_window_size p_slide_size p_LD_threshold \
0 1.0 0.1 200.0 200.0 50.0 0.25
1 1.0 0.1 200.0 200.0 50.0 0.25
2 1.0 0.1 200.0 200.0 50.0 0.25
3 1.0 0.1 200.0 200.0 50.0 0.25
4 1.0 0.1 200.0 200.0 50.0 0.25
5 1.0 0.1 200.0 200.0 50.0 0.25
6 1.0 0.1 200.0 200.0 50.0 0.25
7 1.0 0.1 200.0 200.0 50.0 0.25
8 1.0 0.1 200.0 200.0 50.0 0.25
9 1.0 0.1 200.0 200.0 50.0 0.25
10 1.0 0.1 200.0 200.0 50.0 0.25
11 1.0 0.1 200.0 200.0 50.0 0.25
12 1.0 0.1 200.0 200.0 50.0 0.25
13 1.0 0.1 200.0 200.0 50.0 0.25
14 1.0 0.1 200.0 200.0 50.0 0.25
15 1.0 0.1 200.0 200.0 50.0 0.25
16 1.0 0.1 200.0 200.0 50.0 0.25
17 1.0 0.1 200.0 200.0 50.0 0.25
18 1.0 0.1 200.0 200.0 50.0 0.25
19 1.0 0.1 200.0 200.0 50.0 0.25
pvalue numberofpca tempalpha l1weight ... h2 lambda \
0 1.000000e-10 6.0 0.1 0.1 ... 0.0 0.0
1 3.359818e-10 6.0 0.1 0.1 ... 0.0 0.0
2 1.128838e-09 6.0 0.1 0.1 ... 0.0 0.0
3 3.792690e-09 6.0 0.1 0.1 ... 0.0 0.0
4 1.274275e-08 6.0 0.1 0.1 ... 0.0 0.0
5 4.281332e-08 6.0 0.1 0.1 ... 0.0 0.0
6 1.438450e-07 6.0 0.1 0.1 ... 0.0 0.0
7 4.832930e-07 6.0 0.1 0.1 ... 0.0 0.0
8 1.623777e-06 6.0 0.1 0.1 ... 0.0 0.0
9 5.455595e-06 6.0 0.1 0.1 ... 0.0 0.0
10 1.832981e-05 6.0 0.1 0.1 ... 0.0 0.0
11 6.158482e-05 6.0 0.1 0.1 ... 0.0 0.0
12 2.069138e-04 6.0 0.1 0.1 ... 0.0 0.0
13 6.951928e-04 6.0 0.1 0.1 ... 0.0 0.0
14 2.335721e-03 6.0 0.1 0.1 ... 0.0 0.0
15 7.847600e-03 6.0 0.1 0.1 ... 0.0 0.0
16 2.636651e-02 6.0 0.1 0.1 ... 0.0 0.0
17 8.858668e-02 6.0 0.1 0.1 ... 0.0 0.0
18 2.976351e-01 6.0 0.1 0.1 ... 0.0 0.0
19 1.000000e+00 6.0 0.1 0.1 ... 0.0 0.0
numberofvariants(m) Train_pure_prs Train_null_model Train_best_model \
0 0.0 0.000009 0.23001 0.232329
1 0.0 0.000008 0.23001 0.232706
2 0.0 0.000013 0.23001 0.236337
3 0.0 0.000012 0.23001 0.235599
4 0.0 0.000012 0.23001 0.236509
5 0.0 0.000011 0.23001 0.237139
6 0.0 0.000013 0.23001 0.239885
7 0.0 0.000013 0.23001 0.243846
8 0.0 0.000012 0.23001 0.246030
9 0.0 0.000010 0.23001 0.246566
10 0.0 0.000012 0.23001 0.256612
11 0.0 0.000011 0.23001 0.255474
12 0.0 0.000008 0.23001 0.248666
13 0.0 0.000007 0.23001 0.250900
14 0.0 0.000007 0.23001 0.254301
15 0.0 0.000006 0.23001 0.258285
16 0.0 0.000005 0.23001 0.270212
17 0.0 0.000004 0.23001 0.269347
18 0.0 0.000003 0.23001 0.278149
19 0.0 0.000002 0.23001 0.269891
Test_pure_prs Test_null_model Test_best_model gctb_ld_model
0 -9.758170e-08 0.118692 0.123904 ukbEUR_HM3
1 1.059287e-07 0.118692 0.121482 ukbEUR_HM3
2 2.000056e-06 0.118692 0.125285 ukbEUR_HM3
3 2.674125e-06 0.118692 0.123957 ukbEUR_HM3
4 2.411625e-06 0.118692 0.120808 ukbEUR_HM3
5 2.933871e-06 0.118692 0.123690 ukbEUR_HM3
6 2.668956e-06 0.118692 0.127218 ukbEUR_HM3
7 2.524665e-06 0.118692 0.130111 ukbEUR_HM3
8 2.981676e-06 0.118692 0.130984 ukbEUR_HM3
9 3.506475e-06 0.118692 0.128075 ukbEUR_HM3
10 3.719868e-06 0.118692 0.128822 ukbEUR_HM3
11 3.275230e-06 0.118692 0.132656 ukbEUR_HM3
12 3.477645e-06 0.118692 0.122184 ukbEUR_HM3
13 3.559620e-06 0.118692 0.128199 ukbEUR_HM3
14 3.120129e-06 0.118692 0.130054 ukbEUR_HM3
15 2.821896e-06 0.118692 0.136728 ukbEUR_HM3
16 2.646379e-06 0.118692 0.149808 ukbEUR_HM3
17 2.143497e-06 0.118692 0.158493 ukbEUR_HM3
18 1.520415e-06 0.118692 0.172062 ukbEUR_HM3
19 7.757419e-07 0.118692 0.153734 ukbEUR_HM3
[20 rows x 22 columns]
/tmp/ipykernel_2339320/490051353.py:24: SettingWithCopyWarning:
A value is trying to be set on a copy of a slice from a DataFrame.
Try using .loc[row_indexer,col_indexer] = value instead
See the caveats in the documentation: https://pandas.pydata.org/pandas-docs/stable/user_guide/indexing.html#returning-a-view-versus-a-copy
non_numerical_df[non_numerical_cols] = non_numerical_df[non_numerical_cols].combine_first(df[non_numerical_cols])
/tmp/ipykernel_2339320/490051353.py:24: SettingWithCopyWarning:
A value is trying to be set on a copy of a slice from a DataFrame.
Try using .loc[row_indexer,col_indexer] = value instead
See the caveats in the documentation: https://pandas.pydata.org/pandas-docs/stable/user_guide/indexing.html#returning-a-view-versus-a-copy
non_numerical_df[non_numerical_cols] = non_numerical_df[non_numerical_cols].combine_first(df[non_numerical_cols])
/tmp/ipykernel_2339320/490051353.py:24: SettingWithCopyWarning:
A value is trying to be set on a copy of a slice from a DataFrame.
Try using .loc[row_indexer,col_indexer] = value instead
See the caveats in the documentation: https://pandas.pydata.org/pandas-docs/stable/user_guide/indexing.html#returning-a-view-versus-a-copy
non_numerical_df[non_numerical_cols] = non_numerical_df[non_numerical_cols].combine_first(df[non_numerical_cols])
/tmp/ipykernel_2339320/490051353.py:24: SettingWithCopyWarning:
A value is trying to be set on a copy of a slice from a DataFrame.
Try using .loc[row_indexer,col_indexer] = value instead
See the caveats in the documentation: https://pandas.pydata.org/pandas-docs/stable/user_guide/indexing.html#returning-a-view-versus-a-copy
non_numerical_df[non_numerical_cols] = non_numerical_df[non_numerical_cols].combine_first(df[non_numerical_cols])
Results#
1. Reporting Based on Best Training Performance:#
One can report the results based on the best performance of the training data. For example, if for a specific combination of hyperparameters, the training performance is high, report the corresponding test performance.
Example code:
df = divided_result.sort_values(by='Train_best_model', ascending=False) print(df.iloc[0].to_markdown())
Binary Phenotypes Result Analysis#
You can find the performance quality for binary phenotype using the following template:
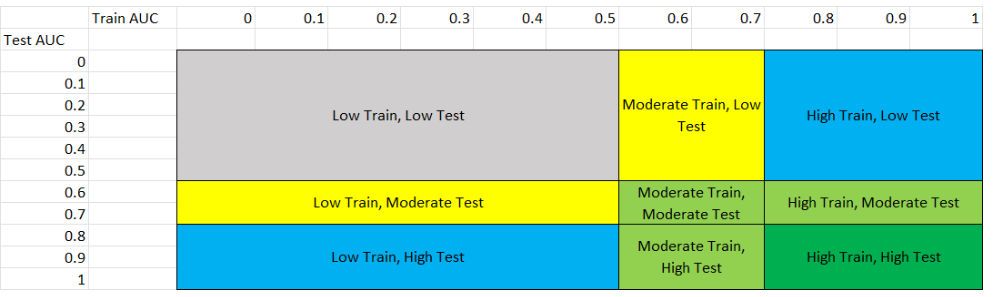
This figure shows the 8 different scenarios that can exist in the results, and the following table explains each scenario.
We classified performance based on the following table:
Performance Level |
Range |
|---|---|
Low Performance |
0 to 0.5 |
Moderate Performance |
0.6 to 0.7 |
High Performance |
0.8 to 1 |
You can match the performance based on the following scenarios:
Scenario |
What’s Happening |
Implication |
|---|---|---|
High Test, High Train |
The model performs well on both training and test datasets, effectively learning the underlying patterns. |
The model is well-tuned, generalizes well, and makes accurate predictions on both datasets. |
High Test, Moderate Train |
The model generalizes well but may not be fully optimized on training data, missing some underlying patterns. |
The model is fairly robust but may benefit from further tuning or more training to improve its learning. |
High Test, Low Train |
An unusual scenario, potentially indicating data leakage or overestimation of test performance. |
The model’s performance is likely unreliable; investigate potential data issues or random noise. |
Moderate Test, High Train |
The model fits the training data well but doesn’t generalize as effectively, capturing only some test patterns. |
The model is slightly overfitting; adjustments may be needed to improve generalization on unseen data. |
Moderate Test, Moderate Train |
The model shows balanced but moderate performance on both datasets, capturing some patterns but missing others. |
The model is moderately fitting; further improvements could be made in both training and generalization. |
Moderate Test, Low Train |
The model underperforms on training data and doesn’t generalize well, leading to moderate test performance. |
The model may need more complexity, additional features, or better training to improve on both datasets. |
Low Test, High Train |
The model overfits the training data, performing poorly on the test set. |
The model doesn’t generalize well; simplifying the model or using regularization may help reduce overfitting. |
Low Test, Low Train |
The model performs poorly on both training and test datasets, failing to learn the data patterns effectively. |
The model is underfitting; it may need more complexity, additional features, or more data to improve performance. |
Recommendations for Publishing Results#
When publishing results, scenarios with moderate train and moderate test performance can be used for complex phenotypes or diseases. However, results showing high train and moderate test, high train and high test, and moderate train and high test are recommended.
For most phenotypes, results typically fall in the moderate train and moderate test performance category.
Continuous Phenotypes Result Analysis#
You can find the performance quality for continuous phenotypes using the following template:
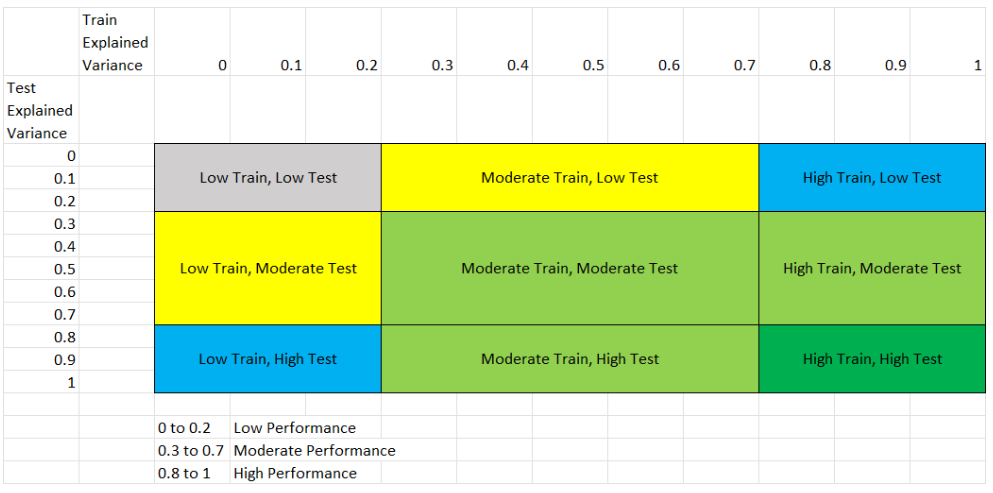
This figure shows the 8 different scenarios that can exist in the results, and the following table explains each scenario.
We classified performance based on the following table:
Performance Level |
Range |
|---|---|
Low Performance |
0 to 0.2 |
Moderate Performance |
0.3 to 0.7 |
High Performance |
0.8 to 1 |
You can match the performance based on the following scenarios:
Scenario |
What’s Happening |
Implication |
|---|---|---|
High Test, High Train |
The model performs well on both training and test datasets, effectively learning the underlying patterns. |
The model is well-tuned, generalizes well, and makes accurate predictions on both datasets. |
High Test, Moderate Train |
The model generalizes well but may not be fully optimized on training data, missing some underlying patterns. |
The model is fairly robust but may benefit from further tuning or more training to improve its learning. |
High Test, Low Train |
An unusual scenario, potentially indicating data leakage or overestimation of test performance. |
The model’s performance is likely unreliable; investigate potential data issues or random noise. |
Moderate Test, High Train |
The model fits the training data well but doesn’t generalize as effectively, capturing only some test patterns. |
The model is slightly overfitting; adjustments may be needed to improve generalization on unseen data. |
Moderate Test, Moderate Train |
The model shows balanced but moderate performance on both datasets, capturing some patterns but missing others. |
The model is moderately fitting; further improvements could be made in both training and generalization. |
Moderate Test, Low Train |
The model underperforms on training data and doesn’t generalize well, leading to moderate test performance. |
The model may need more complexity, additional features, or better training to improve on both datasets. |
Low Test, High Train |
The model overfits the training data, performing poorly on the test set. |
The model doesn’t generalize well; simplifying the model or using regularization may help reduce overfitting. |
Low Test, Low Train |
The model performs poorly on both training and test datasets, failing to learn the data patterns effectively. |
The model is underfitting; it may need more complexity, additional features, or more data to improve performance. |
Recommendations for Publishing Results#
When publishing results, scenarios with moderate train and moderate test performance can be used for complex phenotypes or diseases. However, results showing high train and moderate test, high train and high test, and moderate train and high test are recommended.
For most continuous phenotypes, results typically fall in the moderate train and moderate test performance category.
2. Reporting Generalized Performance:#
One can also report the generalized performance by calculating the difference between the training and test performance, and the sum of the test and training performance. Report the result or hyperparameter combination for which the sum is high and the difference is minimal.
Example code:
df = divided_result.copy() df['Difference'] = abs(df['Train_best_model'] - df['Test_best_model']) df['Sum'] = df['Train_best_model'] + df['Test_best_model'] sorted_df = df.sort_values(by=['Sum', 'Difference'], ascending=[False, True]) print(sorted_df.iloc[0].to_markdown())
3. Reporting Hyperparameters Affecting Test and Train Performance:#
Find the hyperparameters that have more than one unique value and calculate their correlation with the following columns to understand how they are affecting the performance of train and test sets:
Train_null_modelTrain_pure_prsTrain_best_modelTest_pure_prsTest_null_modelTest_best_model
4. Other Analysis#
Once you have the results, you can find how hyperparameters affect the model performance.
Analysis, like overfitting and underfitting, can be performed as well.
The way you are going to report the results can vary.
Results can be visualized, and other patterns in the data can be explored.
import pandas as pd
import seaborn as sns
import matplotlib.pyplot as plt
%matplotlib notebook
import matplotlib
import numpy as np
import matplotlib.pyplot as plt
df = divided_result.sort_values(by='Train_best_model', ascending=False)
print("1. Reporting Based on Best Training Performance:\n")
print(df.iloc[0].to_markdown())
df = divided_result.copy()
# Plot Train and Test best models against p-values
plt.figure(figsize=(10, 6))
plt.plot(df['pvalue'], df['Train_best_model'], label='Train_best_model', marker='o', color='royalblue')
plt.plot(df['pvalue'], df['Test_best_model'], label='Test_best_model', marker='o', color='darkorange')
# Highlight the p-value where both train and test are high
best_index = df[['Train_best_model']].sum(axis=1).idxmax()
best_pvalue = df.loc[best_index, 'pvalue']
best_train = df.loc[best_index, 'Train_best_model']
best_test = df.loc[best_index, 'Test_best_model']
# Use dark colors for the circles
plt.scatter(best_pvalue, best_train, color='darkred', s=100, label=f'Best Performance (Train)', edgecolor='black', zorder=5)
plt.scatter(best_pvalue, best_test, color='darkblue', s=100, label=f'Best Performance (Test)', edgecolor='black', zorder=5)
# Annotate the best performance with p-value, train, and test values
plt.text(best_pvalue, best_train, f'p={best_pvalue:.4g}\nTrain={best_train:.4g}', ha='right', va='bottom', fontsize=9, color='darkred')
plt.text(best_pvalue, best_test, f'p={best_pvalue:.4g}\nTest={best_test:.4g}', ha='right', va='top', fontsize=9, color='darkblue')
# Calculate Difference and Sum
df['Difference'] = abs(df['Train_best_model'] - df['Test_best_model'])
df['Sum'] = df['Train_best_model'] + df['Test_best_model']
# Sort the DataFrame
sorted_df = df.sort_values(by=['Sum', 'Difference'], ascending=[False, True])
#sorted_df = df.sort_values(by=[ 'Difference','Sum'], ascending=[ True,False])
# Highlight the general performance
general_index = sorted_df.index[0]
general_pvalue = sorted_df.loc[general_index, 'pvalue']
general_train = sorted_df.loc[general_index, 'Train_best_model']
general_test = sorted_df.loc[general_index, 'Test_best_model']
plt.scatter(general_pvalue, general_train, color='darkgreen', s=150, label='General Performance (Train)', edgecolor='black', zorder=6)
plt.scatter(general_pvalue, general_test, color='darkorange', s=150, label='General Performance (Test)', edgecolor='black', zorder=6)
# Annotate the general performance with p-value, train, and test values
plt.text(general_pvalue, general_train, f'p={general_pvalue:.4g}\nTrain={general_train:.4g}', ha='left', va='bottom', fontsize=9, color='darkgreen')
plt.text(general_pvalue, general_test, f'p={general_pvalue:.4g}\nTest={general_test:.4g}', ha='left', va='top', fontsize=9, color='darkorange')
# Add labels and legend
plt.xlabel('p-value')
plt.ylabel('Model Performance')
plt.title('Train vs Test Best Models')
plt.legend()
plt.show()
print("2. Reporting Generalized Performance:\n")
df = divided_result.copy()
df['Difference'] = abs(df['Train_best_model'] - df['Test_best_model'])
df['Sum'] = df['Train_best_model'] + df['Test_best_model']
sorted_df = df.sort_values(by=['Sum', 'Difference'], ascending=[False, True])
print(sorted_df.iloc[0].to_markdown())
print("3. Reporting the correlation of hyperparameters and the performance of 'Train_null_model', 'Train_pure_prs', 'Train_best_model', 'Test_pure_prs', 'Test_null_model', and 'Test_best_model':\n")
print("3. For string hyperparameters, we used one-hot encoding to find the correlation between string hyperparameters and 'Train_null_model', 'Train_pure_prs', 'Train_best_model', 'Test_pure_prs', 'Test_null_model', and 'Test_best_model'.")
print("3. We performed this analysis for those hyperparameters that have more than one unique value.")
correlation_columns = [
'Train_null_model', 'Train_pure_prs', 'Train_best_model',
'Test_pure_prs', 'Test_null_model', 'Test_best_model'
]
hyperparams = [col for col in divided_result.columns if len(divided_result[col].unique()) > 1]
hyperparams = list(set(hyperparams+correlation_columns))
# Separate numeric and string columns
numeric_hyperparams = [col for col in hyperparams if pd.api.types.is_numeric_dtype(divided_result[col])]
string_hyperparams = [col for col in hyperparams if pd.api.types.is_string_dtype(divided_result[col])]
# Encode string columns using one-hot encoding
divided_result_encoded = pd.get_dummies(divided_result, columns=string_hyperparams)
# Combine numeric hyperparams with the new one-hot encoded columns
encoded_columns = [col for col in divided_result_encoded.columns if col.startswith(tuple(string_hyperparams))]
hyperparams = numeric_hyperparams + encoded_columns
# Calculate correlations
correlations = divided_result_encoded[hyperparams].corr()
# Display correlation of hyperparameters with train/test performance columns
hyperparam_correlations = correlations.loc[hyperparams, correlation_columns]
hyperparam_correlations = hyperparam_correlations.fillna(0)
# Plotting the correlation heatmap
plt.figure(figsize=(12, 8))
ax = sns.heatmap(hyperparam_correlations, annot=True, cmap='viridis', fmt='.2f', cbar=True)
ax.set_xticklabels(ax.get_xticklabels(), rotation=90, ha='right')
# Rotate y-axis labels to horizontal
#ax.set_yticklabels(ax.get_yticklabels(), rotation=0, va='center')
plt.title('Correlation of Hyperparameters with Train/Test Performance')
plt.show()
sns.set_theme(style="whitegrid") # Choose your preferred style
pairplot = sns.pairplot(divided_result_encoded[hyperparams],hue = 'Test_best_model', palette='viridis')
# Adjust the figure size
pairplot.fig.set_size_inches(15, 15) # You can adjust the size as needed
for ax in pairplot.axes.flatten():
ax.set_xlabel(ax.get_xlabel(), rotation=90, ha='right') # X-axis labels vertical
#ax.set_ylabel(ax.get_ylabel(), rotation=0, va='bottom') # Y-axis labels horizontal
# Show the plot
plt.show()
1. Reporting Based on Best Training Performance:
| | 18 |
|:--------------------|:-----------------------|
| clump_p1 | 1.0 |
| clump_r2 | 0.1 |
| clump_kb | 200.0 |
| p_window_size | 200.0 |
| p_slide_size | 50.0 |
| p_LD_threshold | 0.25 |
| pvalue | 0.2976351441631313 |
| numberofpca | 6.0 |
| tempalpha | 0.1 |
| l1weight | 0.1 |
| bayesmodel | 0.0 |
| model | 0.0 |
| h2 | 0.0 |
| lambda | 0.0 |
| numberofvariants(m) | 0.0 |
| Train_pure_prs | 3.3996311604278516e-06 |
| Train_null_model | 0.23001030414198947 |
| Train_best_model | 0.278149254066261 |
| Test_pure_prs | 1.5204146321723399e-06 |
| Test_null_model | 0.11869244971793831 |
| Test_best_model | 0.17206208984458776 |
| gctb_ld_model | ukbEUR_HM3 |

2. Reporting Generalized Performance:
| | 18 |
|:--------------------|:-----------------------|
| clump_p1 | 1.0 |
| clump_r2 | 0.1 |
| clump_kb | 200.0 |
| p_window_size | 200.0 |
| p_slide_size | 50.0 |
| p_LD_threshold | 0.25 |
| pvalue | 0.2976351441631313 |
| numberofpca | 6.0 |
| tempalpha | 0.1 |
| l1weight | 0.1 |
| bayesmodel | 0.0 |
| model | 0.0 |
| h2 | 0.0 |
| lambda | 0.0 |
| numberofvariants(m) | 0.0 |
| Train_pure_prs | 3.3996311604278516e-06 |
| Train_null_model | 0.23001030414198947 |
| Train_best_model | 0.278149254066261 |
| Test_pure_prs | 1.5204146321723399e-06 |
| Test_null_model | 0.11869244971793831 |
| Test_best_model | 0.17206208984458776 |
| gctb_ld_model | ukbEUR_HM3 |
| Difference | 0.10608716422167325 |
| Sum | 0.45021134391084877 |
3. Reporting the correlation of hyperparameters and the performance of 'Train_null_model', 'Train_pure_prs', 'Train_best_model', 'Test_pure_prs', 'Test_null_model', and 'Test_best_model':
3. For string hyperparameters, we used one-hot encoding to find the correlation between string hyperparameters and 'Train_null_model', 'Train_pure_prs', 'Train_best_model', 'Test_pure_prs', 'Test_null_model', and 'Test_best_model'.
3. We performed this analysis for those hyperparameters that have more than one unique value.

