PRScsx#
In this notebook, we will use PRScsx to calculate the PRS.
Please refer to the original documentation as it is very well written.
PRScsx integrates GWAS summary statistics and external LD reference panels from multiple populations to improve cross-population polygenic prediction. Posterior SNP effect sizes are inferred under coupled continuous shrinkage (CS) priors across populations: PRScsx GitHub.
Download PRScsx#
One can download the PRScs using the following command:
git clone https://github.com/getian107/PRScsx.git
LD Reference Panels#
It uses information from multiple population, so you may need to download the reference panel for multiple population.
1000 Genomes Project Phase 3 Samples#
Population |
Download Link |
Size |
Extraction Command |
|---|---|---|---|
AFR |
~4.44G |
|
|
AMR |
~3.84G |
|
|
EAS |
~4.33G |
|
|
EUR |
~4.56G |
|
|
SAS |
~5.60G |
|
UK Biobank Data (Notes)#
Population |
Download Link |
Size |
Extraction Command |
|---|---|---|---|
AFR |
~4.93G |
|
|
AMR |
~4.10G |
|
|
EAS |
~5.80G |
|
|
EUR |
~6.25G |
|
|
SAS |
~7.37G |
|
For regions that don’t have access to Dropbox, reference panels can be downloaded from the alternative download site.
Download the SNP information file and place it in the same folder containing the reference panels:
1000 Genomes reference: SNP info (~106M)
Download LinkUK Biobank reference: SNP info (~108M)
Download Link
Note: Create a folder PRScsx_reference_panel and save all downloaded datasets in that folder.
If you only download the data for the EUR population, the directory PRScsx_reference_panel would look like this:
.
├── ldblk_1kg_eur
│ ├── ldblk_1kg_chr10.hdf5
│ ├── ldblk_1kg_chr11.hdf5
│ ├── ldblk_1kg_chr12.hdf5
│ ├── ldblk_1kg_chr13.hdf5
│ ├── ldblk_1kg_chr14.hdf5
│ ├── ldblk_1kg_chr15.hdf5
│ ├── ldblk_1kg_chr16.hdf5
│ ├── ldblk_1kg_chr17.hdf5
│ ├── ldblk_1kg_chr18.hdf5
│ ├── ldblk_1kg_chr19.hdf5
│ ├── ldblk_1kg_chr1.hdf5
│ ├── ldblk_1kg_chr20.hdf5
│ ├── ldblk_1kg_chr21.hdf5
│ ├── ldblk_1kg_chr22.hdf5
│ ├── ldblk_1kg_chr2.hdf5
│ ├── ldblk_1kg_chr3.hdf5
│ ├── ldblk_1kg_chr4.hdf5
│ ├── ldblk_1kg_chr5.hdf5
│ ├── ldblk_1kg_chr6.hdf5
│ ├── ldblk_1kg_chr7.hdf5
│ ├── ldblk_1kg_chr8.hdf5
│ ├── ldblk_1kg_chr9.hdf5
│ ├── snpinfo_1kg_hm3
│ └── snpinfo_mult_1kg_hm3
├── ldblk_ukbb_eur
│ ├── ldblk_ukbb_chr10.hdf5
│ ├── ldblk_ukbb_chr11.hdf5
│ ├── ldblk_ukbb_chr12.hdf5
│ ├── ldblk_ukbb_chr13.hdf5
│ ├── ldblk_ukbb_chr14.hdf5
│ ├── ldblk_ukbb_chr15.hdf5
│ ├── ldblk_ukbb_chr16.hdf5
│ ├── ldblk_ukbb_chr17.hdf5
│ ├── ldblk_ukbb_chr18.hdf5
│ ├── ldblk_ukbb_chr19.hdf5
│ ├── ldblk_ukbb_chr1.hdf5
│ ├── ldblk_ukbb_chr20.hdf5
│ ├── ldblk_ukbb_chr21.hdf5
│ ├── ldblk_ukbb_chr22.hdf5
│ ├── ldblk_ukbb_chr2.hdf5
│ ├── ldblk_ukbb_chr3.hdf5
│ ├── ldblk_ukbb_chr4.hdf5
│ ├── ldblk_ukbb_chr5.hdf5
│ ├── ldblk_ukbb_chr6.hdf5
│ ├── ldblk_ukbb_chr7.hdf5
│ ├── ldblk_ukbb_chr8.hdf5
│ ├── ldblk_ukbb_chr9.hdf5
│ ├── snpinfo_mult_ukbb_hm3
│ └── snpinfo_ukbb_hm3
├── snpinfo_mult_1kg_hm3
└── snpinfo_mult_ukbb_hm3
PRScsx Hyperparameters#
PRScsx Parameters#
Parameter |
Description |
Default |
|---|---|---|
|
Sample size of the GWAS. (required) |
N/A |
|
Output directory and output filename prefix of the posterior effect size estimates. (required) |
N/A |
|
Parameter a in the gamma-gamma prior. (optional) |
1 |
|
Parameter b in the gamma-gamma prior. (optional) |
0.5 |
|
Global shrinkage parameter phi. (optional) If not specified, it is learnt from the data using a fully Bayesian approach. |
N/A |
|
Total number of MCMC iterations. (optional) |
1000 |
|
Number of burn-in iterations. (optional) |
500 |
|
Thinning factor of the Markov chain. (optional) |
5 |
|
Chromosome(s) on which the model is fitted, separated by comma (optional). |
1-22 |
|
If True, return standardized posterior SNP effect sizes. If False, return per-allele posterior SNP effect sizes. (optional) |
False |
|
If True, write variant-specific shrinkage estimates. (optional) |
False |
|
If True, write all posterior samples of SNP effect sizes after thinning. (optional) |
False |
PRScs Parameters We Considered and Automated#
We automated the following parameters:
ref_dirs:
["ldblk_1kg_eur", "ldblk_ukbb_eur"]phis:
['auto', '1e-6', '1e-4', '1e-2']valuea:
['1']valueb:
['0.5']number_of_iterations:
['1000']burning_iterations:
['500']thining_iterations:
['5']
GWAS File Processing for PRScsx for Binary Phenotypes#
When the effect size relates to disease risk and is thus given as an odds ratio (OR) rather than BETA (for continuous traits), the PRS is computed as a product of ORs. To simplify this calculation, take the natural logarithm of the OR so that the PRS can be computed using summation instead.
Example Data:#
Using BETA:
SNP |
A1 |
A2 |
BETA |
SE |
|---|---|---|---|---|
rs4970383 |
C |
A |
-0.0064 |
0.0090 |
rs4475691 |
C |
T |
-0.0145 |
0.0094 |
rs13302982 |
A |
G |
-0.0232 |
0.0199 |
Using OR:
SNP |
A1 |
A2 |
OR |
SE |
|---|---|---|---|---|
rs4970383 |
A |
C |
0.9825 |
0.0314 |
rs4475691 |
T |
C |
0.9436 |
0.0319 |
rs13302982 |
A |
G |
1.1337 |
0.0543 |
Multiple population#
python PRScsx.py \
--ref_dir=PRScsx_reference_panel/ldblk_1kg_eur \
--bim_prefix=path_to_bim/test \
--sst_file=path_to_sumstats/EUR_sumstats.txt,path_to_sumstats/EAS_sumstats.txt \
--n_gwas=200000,100000 \
--pop=EUR,EAS \
--chrom=22 \
--phi=1e-2 \
--out_dir=path_to_output \
--out_name=test
Note: We tried to pass multiple reference panels, but it did not work. Therefore, I am assuming that though you can pass the GWAS data for multiple populations, the reference panel should be for one population only (assuming target data).
Note: When using PRScsx, you need to provide multiple GWAS files, the number of samples in GWAS, and the population tag. The reference panel directory should be PRScsx_reference_panel, as created above.
In our case, we considered the same GWAS, reference panel, and population for illustrative purposes.
--sst_file=path_to_sumstats/EUR_sumstats.txt,path_to_sumstats/EAS_sumstats.txt \
--n_gwas=200000,100000 \
--pop=EUR,EAS
import os
import pandas as pd
import numpy as np
import sys
#filedirec = sys.argv[1]
filedirec = "SampleData1"
#filedirec = "asthma_19"
#filedirec = "migraine_0"
# Read the GWAS file.
GWAS = filedirec + os.sep + filedirec+".gz"
df = pd.read_csv(GWAS,compression= "gzip",sep="\s+")
def check_phenotype_is_binary_or_continous(filedirec):
# Read the processed quality controlled file for a phenotype
df = pd.read_csv(filedirec+os.sep+filedirec+'_QC.fam',sep="\s+",header=None)
column_values = df[5].unique()
if len(set(column_values)) == 2:
return "Binary"
else:
return "Continous"
if check_phenotype_is_binary_or_continous(filedirec)=="Binary":
if "BETA" in df.columns.to_list():
# For Binary Phenotypes.
df["OR"] = np.exp(df["BETA"])
df = df[['CHR', 'BP', 'SNP', 'A1', 'A2', 'N', 'SE', 'P', 'OR', 'INFO', 'MAF']]
else:
# For Binary Phenotype.
df = df[['CHR', 'BP', 'SNP', 'A1', 'A2', 'N', 'SE', 'P', 'OR', 'INFO', 'MAF']]
df_transformed = pd.DataFrame({
'SNP': df['SNP'],
'A1': df['A1'],
'A2': df['A2'],
'OR': df['OR'],
'P': df['P'],
})
elif check_phenotype_is_binary_or_continous(filedirec)=="Continous":
if "BETA" in df.columns.to_list():
# For Continous Phenotype.
df = df[['CHR', 'BP', 'SNP', 'A1', 'A2', 'N', 'SE', 'P', 'BETA', 'INFO', 'MAF']]
else:
df["BETA"] = np.log(df["OR"])
df = df[['CHR', 'BP', 'SNP', 'A1', 'A2', 'N', 'SE', 'P', 'BETA', 'INFO', 'MAF']]
df_transformed = pd.DataFrame({
'SNP': df['SNP'],
'A1': df['A1'],
'A2': df['A2'],
'BETA': df['BETA'],
'P': df['P'],
})
n_gwas = df["N"].mean()
df_transformed.to_csv(filedirec + os.sep +filedirec+".PRSCSx",sep="\t",index=False)
print(df_transformed.head().to_markdown())
print("Length of DataFrame!",len(df_transformed))
| | SNP | A1 | A2 | BETA | P |
|---:|:-----------|:-----|:-----|------------:|---------:|
| 0 | rs3131962 | A | G | -0.00211532 | 0.483171 |
| 1 | rs12562034 | A | G | 0.00068708 | 0.834808 |
| 2 | rs4040617 | G | A | -0.00239932 | 0.42897 |
| 3 | rs79373928 | G | T | 0.00203363 | 0.808999 |
| 4 | rs11240779 | G | A | 0.00130747 | 0.590265 |
Length of DataFrame! 499617
Define Hyperparameters#
Define hyperparameters to be optimized and set initial values.
Extract Valid SNPs from Clumped File#
For Windows, download gwak, and for Linux, the awk command is sufficient. For Windows, GWAK is required. You can download it from here. Get it and place it in the same directory.
Execution Path#
At this stage, we have the genotype training data newtrainfilename = "train_data.QC" and genotype test data newtestfilename = "test_data.QC".
We modified the following variables:
filedirec = "SampleData1"orfiledirec = sys.argv[1]foldnumber = "0"orfoldnumber = sys.argv[2]for HPC.
Only these two variables can be modified to execute the code for specific data and specific folds. Though the code can be executed separately for each fold on HPC and separately for each dataset, it is recommended to execute it for multiple diseases and one fold at a time. Here’s the corrected text in Markdown format:
P-values#
PRS calculation relies on P-values. SNPs with low P-values, indicating a high degree of association with a specific trait, are considered for calculation.
You can modify the code below to consider a specific set of P-values and save the file in the same format.
We considered the following parameters:
Minimum P-value:
1e-10Maximum P-value:
1.0Minimum exponent:
10(Minimum P-value in exponent)Number of intervals:
100(Number of intervals to be considered)
The code generates an array of logarithmically spaced P-values:
import numpy as np
import os
minimumpvalue = 10 # Minimum exponent for P-values
numberofintervals = 100 # Number of intervals to be considered
allpvalues = np.logspace(-minimumpvalue, 0, numberofintervals, endpoint=True) # Generating an array of logarithmically spaced P-values
print("Minimum P-value:", allpvalues[0])
print("Maximum P-value:", allpvalues[-1])
count = 1
with open(os.path.join(folddirec, 'range_list'), 'w') as file:
for value in allpvalues:
file.write(f'pv_{value} 0 {value}\n') # Writing range information to the 'range_list' file
count += 1
pvaluefile = os.path.join(folddirec, 'range_list')
In this code:
minimumpvaluedefines the minimum exponent for P-values.numberofintervalsspecifies how many intervals to consider.allpvaluesgenerates an array of P-values spaced logarithmically.The script writes these P-values to a file named
range_listin the specified directory.
from operator import index
import pandas as pd
import numpy as np
import os
import subprocess
import sys
import pandas as pd
import statsmodels.api as sm
import pandas as pd
from sklearn.metrics import roc_auc_score, confusion_matrix
from statsmodels.stats.contingency_tables import mcnemar
def create_directory(directory):
"""Function to create a directory if it doesn't exist."""
if not os.path.exists(directory): # Checking if the directory doesn't exist
os.makedirs(directory) # Creating the directory if it doesn't exist
return directory # Returning the created or existing directory
#foldnumber = sys.argv[1]
foldnumber = "0" # Setting 'foldnumber' to "0"
folddirec = filedirec + os.sep + "Fold_" + foldnumber # Creating a directory path for the specific fold
trainfilename = "train_data" # Setting the name of the training data file
newtrainfilename = "train_data.QC" # Setting the name of the new training data file
testfilename = "test_data" # Setting the name of the test data file
newtestfilename = "test_data.QC" # Setting the name of the new test data file
# Number of PCA to be included as a covariate.
numberofpca = ["6"] # Setting the number of PCA components to be included
# Clumping parameters.
clump_p1 = [1] # List containing clump parameter 'p1'
clump_r2 = [0.1] # List containing clump parameter 'r2'
clump_kb = [200] # List containing clump parameter 'kb'
# Pruning parameters.
p_window_size = [200] # List containing pruning parameter 'window_size'
p_slide_size = [50] # List containing pruning parameter 'slide_size'
p_LD_threshold = [0.25] # List containing pruning parameter 'LD_threshold'
# Kindly note that the number of p-values to be considered varies, and the actual p-value depends on the dataset as well.
# We will specify the range list here.
minimumpvalue = 10 # Minimum p-value in exponent
numberofintervals = 20 # Number of intervals to be considered
allpvalues = np.logspace(-minimumpvalue, 0, numberofintervals, endpoint=True) # Generating an array of logarithmically spaced p-values
count = 1
with open(folddirec + os.sep + 'range_list', 'w') as file:
for value in allpvalues:
file.write(f'pv_{value} 0 {value}\n') # Writing range information to the 'range_list' file
count = count + 1
pvaluefile = folddirec + os.sep + 'range_list'
# Initializing an empty DataFrame with specified column names
prs_result = pd.DataFrame(columns=["clump_p1", "clump_r2", "clump_kb", "p_window_size", "p_slide_size", "p_LD_threshold",
"pvalue", "numberofpca","numberofvariants","Train_pure_prs", "Train_null_model", "Train_best_model",
"Test_pure_prs", "Test_null_model", "Test_best_model"])
Define Helper Functions#
Perform Clumping and Pruning
Calculate PCA Using Plink
Fit Binary Phenotype and Save Results
Fit Continuous Phenotype and Save Results
import os
import subprocess
import pandas as pd
import statsmodels.api as sm
from sklearn.metrics import explained_variance_score
def perform_clumping_and_pruning_on_individual_data(traindirec, newtrainfilename,numberofpca, p1_val, p2_val, p3_val, c1_val, c2_val, c3_val,Name,pvaluefile):
command = [
"./plink",
"--bfile", traindirec+os.sep+newtrainfilename,
"--indep-pairwise", p1_val, p2_val, p3_val,
"--out", traindirec+os.sep+trainfilename
]
subprocess.run(command)
# First perform pruning and then clumping and the pruning.
command = [
"./plink",
"--bfile", traindirec+os.sep+newtrainfilename,
"--clump-p1", c1_val,
"--extract", traindirec+os.sep+trainfilename+".prune.in",
"--clump-r2", c2_val,
"--clump-kb", c3_val,
"--clump", filedirec+os.sep+filedirec+".txt",
"--clump-snp-field", "SNP",
"--clump-field", "P",
"--out", traindirec+os.sep+trainfilename
]
subprocess.run(command)
# Extract the valid SNPs from th clumped file.
# For windows download gwak for linux awk commmand is sufficient.
### For windows require GWAK.
### https://sourceforge.net/projects/gnuwin32/
##3 Get it and place it in the same direc.
#os.system("gawk "+"\""+"NR!=1{print $3}"+"\" "+ traindirec+os.sep+trainfilename+".clumped > "+traindirec+os.sep+trainfilename+".valid.snp")
#print("gawk "+"\""+"NR!=1{print $3}"+"\" "+ traindirec+os.sep+trainfilename+".clumped > "+traindirec+os.sep+trainfilename+".valid.snp")
#Linux:
os.system("awk "+"\""+"NR!=1{print $3}"+"\" "+ traindirec+os.sep+trainfilename+".clumped > "+traindirec+os.sep+trainfilename+".valid.snp")
#print("awk "+"\""+"NR!=1{print $3}"+"\" "+ traindirec+os.sep+trainfilename+".clumped > "+traindirec+os.sep+trainfilename+".valid.snp")
command = [
"./plink",
"--make-bed",
"--bfile", traindirec+os.sep+newtrainfilename,
"--indep-pairwise", p1_val, p2_val, p3_val,
"--extract", traindirec+os.sep+trainfilename+".valid.snp",
"--out", traindirec+os.sep+newtrainfilename+".clumped.pruned"
]
subprocess.run(command)
command = [
"./plink",
"--make-bed",
"--bfile", traindirec+os.sep+testfilename,
"--indep-pairwise", p1_val, p2_val, p3_val,
"--extract", traindirec+os.sep+trainfilename+".valid.snp",
"--out", traindirec+os.sep+testfilename+".clumped.pruned"
]
subprocess.run(command)
def calculate_pca_for_traindata_testdata_for_clumped_pruned_snps(traindirec, newtrainfilename,p):
# Calculate the PRS for the test data using the same set of SNPs and also calculate the PCA.
# Also extract the PCA at this point.
# PCA are calculated afer clumping and pruining.
command = [
"./plink",
"--bfile", folddirec+os.sep+testfilename+".clumped.pruned",
# Select the final variants after clumping and pruning.
"--extract", traindirec+os.sep+trainfilename+".valid.snp",
"--pca", p,
"--out", folddirec+os.sep+testfilename
]
subprocess.run(command)
command = [
"./plink",
"--bfile", traindirec+os.sep+newtrainfilename+".clumped.pruned",
# Select the final variants after clumping and pruning.
"--extract", traindirec+os.sep+trainfilename+".valid.snp",
"--pca", p,
"--out", traindirec+os.sep+trainfilename
]
subprocess.run(command)
# This function fit the binary model on the PRS.
def fit_binary_phenotype_on_PRS(traindirec, newtrainfilename, p,ref_dir,phi,va,vb,number_of_iteration,burning_iteration,thining_iteration, p1_val, p2_val, p3_val, c1_val, c2_val, c3_val,Name,pvaluefile):
threshold_values = allpvalues
# Merge the covariates, pca and phenotypes.
tempphenotype_train = pd.read_table(traindirec+os.sep+newtrainfilename+".clumped.pruned"+".fam", sep="\s+",header=None)
phenotype_train = pd.DataFrame()
phenotype_train["Phenotype"] = tempphenotype_train[5].values
pcs_train = pd.read_table(traindirec+os.sep+trainfilename+".eigenvec", sep="\s+",header=None, names=["FID", "IID"] + [f"PC{str(i)}" for i in range(1, int(p)+1)])
covariate_train = pd.read_table(traindirec+os.sep+trainfilename+".cov",sep="\s+")
covariate_train.fillna(0, inplace=True)
covariate_train = covariate_train[covariate_train["FID"].isin(pcs_train["FID"].values) & covariate_train["IID"].isin(pcs_train["IID"].values)]
covariate_train['FID'] = covariate_train['FID'].astype(str)
pcs_train['FID'] = pcs_train['FID'].astype(str)
covariate_train['IID'] = covariate_train['IID'].astype(str)
pcs_train['IID'] = pcs_train['IID'].astype(str)
covandpcs_train = pd.merge(covariate_train, pcs_train, on=["FID","IID"])
covandpcs_train.fillna(0, inplace=True)
## Scale the covariates!
from sklearn.preprocessing import MinMaxScaler
from sklearn.metrics import explained_variance_score
scaler = MinMaxScaler()
normalized_values_train = scaler.fit_transform(covandpcs_train.iloc[:, 2:])
#covandpcs_train.iloc[:, 2:] = normalized_values_test
tempphenotype_test = pd.read_table(traindirec+os.sep+testfilename+".clumped.pruned"+".fam", sep="\s+",header=None)
phenotype_test= pd.DataFrame()
phenotype_test["Phenotype"] = tempphenotype_test[5].values
pcs_test = pd.read_table(traindirec+os.sep+testfilename+".eigenvec", sep="\s+",header=None, names=["FID", "IID"] + [f"PC{str(i)}" for i in range(1, int(p)+1)])
covariate_test = pd.read_table(traindirec+os.sep+testfilename+".cov",sep="\s+")
covariate_test.fillna(0, inplace=True)
covariate_test = covariate_test[covariate_test["FID"].isin(pcs_test["FID"].values) & covariate_test["IID"].isin(pcs_test["IID"].values)]
covariate_test['FID'] = covariate_test['FID'].astype(str)
pcs_test['FID'] = pcs_test['FID'].astype(str)
covariate_test['IID'] = covariate_test['IID'].astype(str)
pcs_test['IID'] = pcs_test['IID'].astype(str)
covandpcs_test = pd.merge(covariate_test, pcs_test, on=["FID","IID"])
covandpcs_test.fillna(0, inplace=True)
normalized_values_test = scaler.transform(covandpcs_test.iloc[:, 2:])
#covandpcs_test.iloc[:, 2:] = normalized_values_test
tempalphas = [0.1,0.2,0.3,0.4,0.5,0.6,0.7,0.8,0.9]
l1weights = [0.1,0.2,0.3,0.4,0.5,0.6,0.7,0.8,0.9]
tempalphas = [0.1]
l1weights = [0.1]
phenotype_train["Phenotype"] = phenotype_train["Phenotype"].replace({1: 0, 2: 1})
phenotype_test["Phenotype"] = phenotype_test["Phenotype"].replace({1: 0, 2: 1})
for tempalpha in tempalphas:
for l1weight in l1weights:
try:
null_model = sm.Logit(phenotype_train["Phenotype"], sm.add_constant(covandpcs_train.iloc[:, 2:])).fit_regularized(alpha=tempalpha, L1_wt=l1weight)
#null_model = sm.Logit(phenotype_train["Phenotype"], sm.add_constant(covandpcs_train.iloc[:, 2:])).fit()
except:
print("XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX")
continue
train_null_predicted = null_model.predict(sm.add_constant(covandpcs_train.iloc[:, 2:]))
from sklearn.metrics import roc_auc_score, confusion_matrix
from sklearn.metrics import r2_score
test_null_predicted = null_model.predict(sm.add_constant(covandpcs_test.iloc[:, 2:]))
global prs_result
for i in threshold_values:
try:
prs_train = pd.read_table(traindirec+os.sep+Name+os.sep+"train_data.pv_"+f"{i}.profile", sep="\s+", usecols=["FID", "IID", "SCORE"])
except:
continue
prs_train['FID'] = prs_train['FID'].astype(str)
prs_train['IID'] = prs_train['IID'].astype(str)
try:
prs_test = pd.read_table(traindirec+os.sep+Name+os.sep+"test_data.pv_"+f"{i}.profile", sep="\s+", usecols=["FID", "IID", "SCORE"])
except:
continue
prs_test['FID'] = prs_test['FID'].astype(str)
prs_test['IID'] = prs_test['IID'].astype(str)
pheno_prs_train = pd.merge(covandpcs_train, prs_train, on=["FID", "IID"])
pheno_prs_test = pd.merge(covandpcs_test, prs_test, on=["FID", "IID"])
try:
model = sm.Logit(phenotype_train["Phenotype"], sm.add_constant(pheno_prs_train.iloc[:, 2:])).fit_regularized(alpha=tempalpha, L1_wt=l1weight)
#model = sm.Logit(phenotype_train["Phenotype"], sm.add_constant(pheno_prs_train.iloc[:, 2:])).fit()
except:
continue
train_best_predicted = model.predict(sm.add_constant(pheno_prs_train.iloc[:, 2:]))
test_best_predicted = model.predict(sm.add_constant(pheno_prs_test.iloc[:, 2:]))
from sklearn.metrics import roc_auc_score, confusion_matrix
prs_result = prs_result._append({
"clump_p1": c1_val,
"clump_r2": c2_val,
"clump_kb": c3_val,
"p_window_size": p1_val,
"p_slide_size": p2_val,
"p_LD_threshold": p3_val,
"pvalue": i,
"numberofpca":p,
"tempalpha":str(tempalpha),
"l1weight":str(l1weight),
"PRScs_ref_dir":ref_dir,
"PRScs_phi":phi,
"PRScs_va":va,
"PRScs_vb":vb,
"PRScs_number_of_iteration":number_of_iteration,
"PRScs_burning_iteration":burning_iteration,
"PRScs_thining_iteration":thining_iteration,
"Train_pure_prs":roc_auc_score(phenotype_train["Phenotype"].values,prs_train['SCORE'].values),
"Train_null_model":roc_auc_score(phenotype_train["Phenotype"].values,train_null_predicted.values),
"Train_best_model":roc_auc_score(phenotype_train["Phenotype"].values,train_best_predicted.values),
"Test_pure_prs":roc_auc_score(phenotype_test["Phenotype"].values,prs_test['SCORE'].values),
"Test_null_model":roc_auc_score(phenotype_test["Phenotype"].values,test_null_predicted.values),
"Test_best_model":roc_auc_score(phenotype_test["Phenotype"].values,test_best_predicted.values),
}, ignore_index=True)
prs_result.to_csv(traindirec+os.sep+Name+os.sep+"Results.csv",index=False)
return
# This function fit the binary model on the PRS.
def fit_continous_phenotype_on_PRS(traindirec, newtrainfilename, p,ref_dir,phi,va,vb,number_of_iteration,burning_iteration,thining_iteration, p1_val, p2_val, p3_val, c1_val, c2_val, c3_val,Name,pvaluefile):
threshold_values = allpvalues
# Merge the covariates, pca and phenotypes.
tempphenotype_train = pd.read_table(traindirec+os.sep+newtrainfilename+".clumped.pruned"+".fam", sep="\s+",header=None)
phenotype_train = pd.DataFrame()
phenotype_train["Phenotype"] = tempphenotype_train[5].values
pcs_train = pd.read_table(traindirec+os.sep+trainfilename+".eigenvec", sep="\s+",header=None, names=["FID", "IID"] + [f"PC{str(i)}" for i in range(1, int(p)+1)])
covariate_train = pd.read_table(traindirec+os.sep+trainfilename+".cov",sep="\s+")
covariate_train.fillna(0, inplace=True)
covariate_train = covariate_train[covariate_train["FID"].isin(pcs_train["FID"].values) & covariate_train["IID"].isin(pcs_train["IID"].values)]
covariate_train['FID'] = covariate_train['FID'].astype(str)
pcs_train['FID'] = pcs_train['FID'].astype(str)
covariate_train['IID'] = covariate_train['IID'].astype(str)
pcs_train['IID'] = pcs_train['IID'].astype(str)
covandpcs_train = pd.merge(covariate_train, pcs_train, on=["FID","IID"])
covandpcs_train.fillna(0, inplace=True)
## Scale the covariates!
from sklearn.preprocessing import MinMaxScaler
from sklearn.metrics import explained_variance_score
scaler = MinMaxScaler()
normalized_values_train = scaler.fit_transform(covandpcs_train.iloc[:, 2:])
#covandpcs_train.iloc[:, 2:] = normalized_values_test
tempphenotype_test = pd.read_table(traindirec+os.sep+testfilename+".clumped.pruned"+".fam", sep="\s+",header=None)
phenotype_test= pd.DataFrame()
phenotype_test["Phenotype"] = tempphenotype_test[5].values
pcs_test = pd.read_table(traindirec+os.sep+testfilename+".eigenvec", sep="\s+",header=None, names=["FID", "IID"] + [f"PC{str(i)}" for i in range(1, int(p)+1)])
covariate_test = pd.read_table(traindirec+os.sep+testfilename+".cov",sep="\s+")
covariate_test.fillna(0, inplace=True)
covariate_test = covariate_test[covariate_test["FID"].isin(pcs_test["FID"].values) & covariate_test["IID"].isin(pcs_test["IID"].values)]
covariate_test['FID'] = covariate_test['FID'].astype(str)
pcs_test['FID'] = pcs_test['FID'].astype(str)
covariate_test['IID'] = covariate_test['IID'].astype(str)
pcs_test['IID'] = pcs_test['IID'].astype(str)
covandpcs_test = pd.merge(covariate_test, pcs_test, on=["FID","IID"])
covandpcs_test.fillna(0, inplace=True)
normalized_values_test = scaler.transform(covandpcs_test.iloc[:, 2:])
#covandpcs_test.iloc[:, 2:] = normalized_values_test
tempalphas = [0.1,0.2,0.3,0.4,0.5,0.6,0.7,0.8,0.9]
l1weights = [0.1,0.2,0.3,0.4,0.5,0.6,0.7,0.8,0.9]
tempalphas = [0.1]
l1weights = [0.1]
#phenotype_train["Phenotype"] = phenotype_train["Phenotype"].replace({1: 0, 2: 1})
#phenotype_test["Phenotype"] = phenotype_test["Phenotype"].replace({1: 0, 2: 1})
for tempalpha in tempalphas:
for l1weight in l1weights:
try:
#null_model = sm.OLS(phenotype_train["Phenotype"], sm.add_constant(covandpcs_train.iloc[:, 2:])).fit_regularized(alpha=tempalpha, L1_wt=l1weight)
null_model = sm.OLS(phenotype_train["Phenotype"], sm.add_constant(covandpcs_train.iloc[:, 2:])).fit()
#null_model = sm.OLS(phenotype_train["Phenotype"], sm.add_constant(covandpcs_train.iloc[:, 2:])).fit()
except:
print("XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX")
continue
train_null_predicted = null_model.predict(sm.add_constant(covandpcs_train.iloc[:, 2:]))
from sklearn.metrics import roc_auc_score, confusion_matrix
from sklearn.metrics import r2_score
test_null_predicted = null_model.predict(sm.add_constant(covandpcs_test.iloc[:, 2:]))
global prs_result
for i in threshold_values:
try:
prs_train = pd.read_table(traindirec+os.sep+Name+os.sep+"train_data.pv_"+f"{i}.profile", sep="\s+", usecols=["FID", "IID", "SCORE"])
except:
continue
prs_train['FID'] = prs_train['FID'].astype(str)
prs_train['IID'] = prs_train['IID'].astype(str)
try:
prs_test = pd.read_table(traindirec+os.sep+Name+os.sep+"test_data.pv_"+f"{i}.profile", sep="\s+", usecols=["FID", "IID", "SCORE"])
except:
continue
prs_test['FID'] = prs_test['FID'].astype(str)
prs_test['IID'] = prs_test['IID'].astype(str)
pheno_prs_train = pd.merge(covandpcs_train, prs_train, on=["FID", "IID"])
pheno_prs_test = pd.merge(covandpcs_test, prs_test, on=["FID", "IID"])
try:
#model = sm.OLS(phenotype_train["Phenotype"], sm.add_constant(pheno_prs_train.iloc[:, 2:])).fit_regularized(alpha=tempalpha, L1_wt=l1weight)
model = sm.OLS(phenotype_train["Phenotype"], sm.add_constant(pheno_prs_train.iloc[:, 2:])).fit()
except:
continue
train_best_predicted = model.predict(sm.add_constant(pheno_prs_train.iloc[:, 2:]))
test_best_predicted = model.predict(sm.add_constant(pheno_prs_test.iloc[:, 2:]))
from sklearn.metrics import roc_auc_score, confusion_matrix
prs_result = prs_result._append({
"clump_p1": c1_val,
"clump_r2": c2_val,
"clump_kb": c3_val,
"p_window_size": p1_val,
"p_slide_size": p2_val,
"p_LD_threshold": p3_val,
"pvalue": i,
"numberofpca":p,
"tempalpha":str(tempalpha),
"l1weight":str(l1weight),
"PRScs_ref_dir":ref_dir,
"PRScs_phi":phi,
"PRScs_va":va,
"PRScs_vb":vb,
"PRScs_number_of_iteration":number_of_iteration,
"PRScs_burning_iteration":burning_iteration,
"PRScs_thining_iteration":thining_iteration,
"Train_pure_prs":explained_variance_score(phenotype_train["Phenotype"],prs_train['SCORE'].values),
"Train_null_model":explained_variance_score(phenotype_train["Phenotype"],train_null_predicted),
"Train_best_model":explained_variance_score(phenotype_train["Phenotype"],train_best_predicted),
"Test_pure_prs":explained_variance_score(phenotype_test["Phenotype"],prs_test['SCORE'].values),
"Test_null_model":explained_variance_score(phenotype_test["Phenotype"],test_null_predicted),
"Test_best_model":explained_variance_score(phenotype_test["Phenotype"],test_best_predicted),
}, ignore_index=True)
prs_result.to_csv(traindirec+os.sep+Name+os.sep+"Results.csv",index=False)
return
Execute PRScsx#
# Define a global variable to store results
prs_result = pd.DataFrame()
def transform_prscs_data(traindirec, newtrainfilename,numberofpca,ref_dir,phi,va,vb,number_of_iteration,burning_iteration,thining_iteration, p1_val, p2_val, p3_val, c1_val, c2_val, c3_val,Name,pvaluefile):
### First perform clumping on the file and save the clumpled file.
# perform_clumping_and_pruning_on_individual_data(traindirec, newtrainfilename,p, p1_val, p2_val, p3_val, c1_val, c2_val, c3_val,Name,pvaluefile)
#newtrainfilename = newtrainfilename+".clumped.pruned"
#testfilename = testfilename+".clumped.pruned"
#clupmedfile = traindirec+os.sep+newtrainfilename+".clump"
#prunedfile = traindirec+os.sep+newtrainfilename+".clumped.pruned"
# Also extract the PCA at this point for both test and training data.
# calculate_pca_for_traindata_testdata_for_clumped_pruned_snps(traindirec, newtrainfilename,p)
#Extract p-values from the GWAS file.
# Command for Linux.
os.system("awk "+"\'"+"{print $3,$8}"+"\'"+" ./"+filedirec+os.sep+filedirec+".txt > ./"+traindirec+os.sep+"SNP.pvalue")
# Command for windows.
### For windows get GWAK.
### https://sourceforge.net/projects/gnuwin32/
##3 Get it and place it in the same direc.
#os.system("gawk "+"\""+"{print $3,$8}"+"\""+" ./"+filedirec+os.sep+filedirec+".txt > ./"+traindirec+os.sep+"SNP.pvalue")
#print("gawk "+"\""+"{print $3,$8}"+"\""+" ./"+filedirec+os.sep+filedirec+".txt > ./"+traindirec+os.sep+"SNP.pvalue")
#exit(0)
# Delete the files generated in the previous iteration.
import glob
file_list = glob.glob(traindirec+os.sep+"PRSCSx*.txt")
sorted_file_list = sorted(file_list, key=lambda x: int(''.join(filter(str.isdigit, x))))
def delete_files(file_list):
for file in file_list:
try:
os.remove(file)
print(f"File {file} deleted successfully.")
except FileNotFoundError:
print(f"File {file} not found.")
except Exception as e:
print(f"Error deleting file {file}: {e}")
delete_files(file_list)
files_to_remove = [
traindirec+os.sep+".PRSCSx_final_GWAS",
]
# Loop through the files and remove them if they exist
for file_path in files_to_remove:
if os.path.exists(file_path):
os.remove(file_path)
print(f"Removed: {file_path}")
else:
print(f"File does not exist: {file_path}")
path_to_PRScsx_reference_panel = "PRScsx_reference_panel"
if phi=="auto":
command = [
"python", # Use python3 instead of python
"PRScsx/PRScsx.py",
# First and second reference panel seperated by comma.
"--ref_dir=" + path_to_PRScsx_reference_panel+"/"+ref_dir,
# bim file is going to be the same.
"--bim_prefix=" + traindirec+os.sep+newtrainfilename+".clumped.pruned",
# Fist and the second gwas seperated by comma.
"--sst_file=" + filedirec + os.sep +filedirec+".PRSCSx" +","+filedirec + os.sep +filedirec+".PRSCSx",
"--n_gwas=" + str(int(n_gwas))+","+str(int(n_gwas)),
"--pop=" + "EUR"+","+"EUR",
"--a="+str(va),
"--b="+str(vb),
"--n_iter="+str(number_of_iteration),
"--n_burnin="+str(burning_iteration),
"--thin="+str(thining_iteration),
"--out_dir=" + traindirec,
"--out_name="+"PRSCSx"
]
print(" ".join(command))
subprocess.run(command)
else:
command = [
"python", # Use python3 instead of python
"PRScsx/PRScsx.py",
"--ref_dir=" + path_to_PRScsx_reference_panel+"/"+ref_dir,
# bim file is going to be the same.
"--bim_prefix=" + traindirec+os.sep+newtrainfilename+".clumped.pruned",
# Fist and the second gwas seperated by comma.
"--sst_file=" + filedirec + os.sep +filedirec+".PRSCSx" +","+filedirec + os.sep +filedirec+".PRSCSx",
"--n_gwas=" + str(int(n_gwas))+","+str(int(n_gwas)),
"--pop=" + "EUR"+","+"EUR",
"--a="+str(va),
"--b="+str(vb),
"--n_iter="+str(number_of_iteration),
"--n_burnin="+str(burning_iteration),
"--thin="+str(thining_iteration),
"--phi=" + str(phi),
"--out_dir=" + traindirec,
"--out_name="+"PRSCSx"
]
subprocess.run(command)
print(" ".join(command))
import glob
file_list = glob.glob(traindirec+os.sep+"PRSCSx*.txt")
sorted_file_list = sorted(file_list, key=lambda x: int(''.join(filter(str.isdigit, x))))
merged_df = pd.DataFrame()
# Iterate through the files
for file in file_list:
try:
df = pd.read_csv(file, header=None,sep="\s+") # No header
merged_df = pd.concat([merged_df, df], ignore_index=True)
except:
pass
# Reset column names
num_columns = len(merged_df.columns)
merged_df.columns = ["CHR","SNP","BP","A1","A2","NewBeta"]
if check_phenotype_is_binary_or_continous(filedirec)=="Binary":
merged_df["NewBeta"] = np.exp(merged_df["NewBeta"])
else:
pass
merged_df[["SNP","A1","NewBeta"]].to_csv(traindirec+os.sep+".PRSCSx_final_GWAS",sep="\t",index=False)
# Caluclate Plink Score.
command = [
"./plink",
"--bfile", traindirec+os.sep+newtrainfilename,
### SNP column = 3, Effect allele column 1 = 4, OR column=9
"--score", traindirec+os.sep+".PRSCSx_final_GWAS", "1", "2", "3", "header",
"--q-score-range", traindirec+os.sep+"range_list",traindirec+os.sep+"SNP.pvalue",
"--extract", traindirec+os.sep+trainfilename+".valid.snp",
"--out", traindirec+os.sep+Name+os.sep+trainfilename
]
#exit(0)
subprocess.run(command)
# Calculate the PRS for the test data using the same set of SNPs and also calculate the PCA.
command = [
"./plink",
"--bfile", folddirec+os.sep+testfilename,
### SNP column = 3, Effect allele column 1 = 4, OR column=9
"--score", traindirec+os.sep+".PRSCSx_final_GWAS", "1", "2", "3", "header",
"--q-score-range", traindirec+os.sep+"range_list",traindirec+os.sep+"SNP.pvalue",
"--extract", traindirec+os.sep+trainfilename+".valid.snp",
"--out", folddirec+os.sep+Name+os.sep+testfilename
]
subprocess.run(command)
# At this stage the scores are finalizied.
# The next step is to fit the model and find the explained variance by each profile.
# Load the PCA and Load the Covariates for trainingdatafirst.
if check_phenotype_is_binary_or_continous(filedirec)=="Binary":
print("Binary Phenotype!")
fit_binary_phenotype_on_PRS(traindirec, newtrainfilename, p,ref_dir,phi,va,vb,number_of_iteration,burning_iteration,thining_iteration, p1_val, p2_val, p3_val, c1_val, c2_val, c3_val,Name,pvaluefile)
else:
print("Continous Phenotype!")
fit_continous_phenotype_on_PRS(traindirec, newtrainfilename, p,ref_dir,phi,va,vb,number_of_iteration,burning_iteration,thining_iteration, p1_val, p2_val, p3_val, c1_val, c2_val, c3_val,Name,pvaluefile)
ref_dirs = ["ldblk_1kg_eur","ldblk_ukbb_eur"]
phis = ['auto','1e-6', '1e-4', '1e-2',]
phis = ['auto','1e-6']
valuea = ['1']
valueb = ['0.5']
number_of_iterations = ['1000']
burning_iterations = ['500']
thining_iterations = ['5']
result_directory = "PRScsx"
# Nested loops to iterate over different parameter values
create_directory(folddirec+os.sep+result_directory)
for p1_val in p_window_size:
for p2_val in p_slide_size:
for p3_val in p_LD_threshold:
for c1_val in clump_p1:
for c2_val in clump_r2:
for c3_val in clump_kb:
for p in numberofpca:
for ref_dir in ref_dirs:
for phi in phis:
for va in valuea:
for vb in valueb:
for number_of_iteration in number_of_iterations:
for burning_iteration in burning_iterations:
for thining_iteration in thining_iterations:
transform_prscs_data(folddirec, newtrainfilename, p,ref_dir,phi,va,vb,number_of_iteration,burning_iteration,thining_iteration, str(p1_val), str(p2_val), str(p3_val), str(c1_val), str(c2_val), str(c3_val),result_directory, pvaluefile)
python PRScsx/PRScsx.py --ref_dir=PRScsx_reference_panel/ldblk_1kg_eur --bim_prefix=SampleData1/Fold_0/train_data.QC.clumped.pruned --sst_file=SampleData1/SampleData1.PRSCSx,SampleData1/SampleData1.PRSCSx --n_gwas=388028,388028 --pop=EUR,EUR --a=1 --b=0.5 --n_iter=1000 --n_burnin=500 --thin=5 --out_dir=SampleData1/Fold_0 --out_name=PRSCSx
--ref_dir=PRScsx_reference_panel/ldblk_1kg_eur
--bim_prefix=SampleData1/Fold_0/train_data.QC.clumped.pruned
--sst_file=['SampleData1/SampleData1.PRSCSx', 'SampleData1/SampleData1.PRSCSx']
--a=1.0
--b=0.5
--phi=None
--n_gwas=[388028, 388028]
--pop=['EUR', 'EUR']
--n_iter=1000
--n_burnin=500
--thin=5
--out_dir=SampleData1/Fold_0
--out_name=PRSCSx
--chrom=range(1, 23)
--meta=FALSE
--seed=None
*** 2 discovery populations detected ***
##### process chromosome 1 #####
... parse reference file: PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... 109168 SNPs on chromosome 1 read from PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... parse bim file: SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... 14013 SNPs on chromosome 1 read from SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 3103 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 3103 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR reference LD on chromosome 1 ...
... parse EUR reference LD on chromosome 1 ...
... align reference LD on chromosome 1 across populations ...
... 3103 valid SNPs across populations ...
... MCMC ...
--- iter-100 ---
--- iter-200 ---
--- iter-300 ---
--- iter-400 ---
--- iter-500 ---
--- iter-600 ---
--- iter-700 ---
--- iter-800 ---
--- iter-900 ---
--- iter-1000 ---
... Estimated global shrinkage parameter: 7.97e-02 ...
... Done ...
##### process chromosome 2 #####
... parse reference file: PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... 108968 SNPs on chromosome 2 read from PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... parse bim file: SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... 13813 SNPs on chromosome 2 read from SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 2705 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 2705 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR reference LD on chromosome 2 ...
... parse EUR reference LD on chromosome 2 ...
... align reference LD on chromosome 2 across populations ...
... 2705 valid SNPs across populations ...
... MCMC ...
--- iter-100 ---
--- iter-200 ---
--- iter-300 ---
--- iter-400 ---
--- iter-500 ---
--- iter-600 ---
--- iter-700 ---
--- iter-800 ---
--- iter-900 ---
--- iter-1000 ---
... Estimated global shrinkage parameter: 8.25e-02 ...
... Done ...
##### process chromosome 3 #####
... parse reference file: PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... 90368 SNPs on chromosome 3 read from PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... parse bim file: SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... 11785 SNPs on chromosome 3 read from SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 2307 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 2307 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR reference LD on chromosome 3 ...
... parse EUR reference LD on chromosome 3 ...
... align reference LD on chromosome 3 across populations ...
... 2307 valid SNPs across populations ...
... MCMC ...
--- iter-100 ---
--- iter-200 ---
--- iter-300 ---
--- iter-400 ---
--- iter-500 ---
--- iter-600 ---
--- iter-700 ---
--- iter-800 ---
--- iter-900 ---
--- iter-1000 ---
... Estimated global shrinkage parameter: 8.87e-02 ...
... Done ...
##### process chromosome 4 #####
... parse reference file: PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... 80831 SNPs on chromosome 4 read from PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... parse bim file: SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... 11041 SNPs on chromosome 4 read from SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1991 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1991 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR reference LD on chromosome 4 ...
... parse EUR reference LD on chromosome 4 ...
... align reference LD on chromosome 4 across populations ...
... 1991 valid SNPs across populations ...
... MCMC ...
--- iter-100 ---
--- iter-200 ---
--- iter-300 ---
--- iter-400 ---
--- iter-500 ---
--- iter-600 ---
--- iter-700 ---
--- iter-800 ---
--- iter-900 ---
--- iter-1000 ---
... Estimated global shrinkage parameter: 8.58e-02 ...
... Done ...
##### process chromosome 5 #####
... parse reference file: PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... 80843 SNPs on chromosome 5 read from PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... parse bim file: SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... 10632 SNPs on chromosome 5 read from SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 2190 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 2190 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR reference LD on chromosome 5 ...
... parse EUR reference LD on chromosome 5 ...
... align reference LD on chromosome 5 across populations ...
... 2190 valid SNPs across populations ...
... MCMC ...
--- iter-100 ---
--- iter-200 ---
--- iter-300 ---
--- iter-400 ---
--- iter-500 ---
--- iter-600 ---
--- iter-700 ---
--- iter-800 ---
--- iter-900 ---
--- iter-1000 ---
... Estimated global shrinkage parameter: 7.88e-02 ...
... Done ...
##### process chromosome 6 #####
... parse reference file: PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... 85723 SNPs on chromosome 6 read from PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... parse bim file: SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... 10068 SNPs on chromosome 6 read from SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 2280 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 2280 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR reference LD on chromosome 6 ...
... parse EUR reference LD on chromosome 6 ...
... align reference LD on chromosome 6 across populations ...
... 2280 valid SNPs across populations ...
... MCMC ...
--- iter-100 ---
--- iter-200 ---
--- iter-300 ---
--- iter-400 ---
--- iter-500 ---
--- iter-600 ---
--- iter-700 ---
--- iter-800 ---
--- iter-900 ---
--- iter-1000 ---
... Estimated global shrinkage parameter: 1.03e-01 ...
... Done ...
##### process chromosome 7 #####
... parse reference file: PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... 70555 SNPs on chromosome 7 read from PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... parse bim file: SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... 9496 SNPs on chromosome 7 read from SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1865 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1865 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR reference LD on chromosome 7 ...
... parse EUR reference LD on chromosome 7 ...
... align reference LD on chromosome 7 across populations ...
... 1865 valid SNPs across populations ...
... MCMC ...
--- iter-100 ---
--- iter-200 ---
--- iter-300 ---
--- iter-400 ---
--- iter-500 ---
--- iter-600 ---
--- iter-700 ---
--- iter-800 ---
--- iter-900 ---
--- iter-1000 ---
... Estimated global shrinkage parameter: 8.22e-02 ...
... Done ...
##### process chromosome 8 #####
... parse reference file: PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... 69913 SNPs on chromosome 8 read from PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... parse bim file: SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... 8867 SNPs on chromosome 8 read from SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1775 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1775 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR reference LD on chromosome 8 ...
... parse EUR reference LD on chromosome 8 ...
... align reference LD on chromosome 8 across populations ...
... 1775 valid SNPs across populations ...
... MCMC ...
--- iter-100 ---
--- iter-200 ---
--- iter-300 ---
--- iter-400 ---
--- iter-500 ---
--- iter-600 ---
--- iter-700 ---
--- iter-800 ---
--- iter-900 ---
--- iter-1000 ---
... Estimated global shrinkage parameter: 9.84e-02 ...
... Done ...
##### process chromosome 9 #####
... parse reference file: PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... 59066 SNPs on chromosome 9 read from PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... parse bim file: SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... 7768 SNPs on chromosome 9 read from SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1758 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1758 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR reference LD on chromosome 9 ...
... parse EUR reference LD on chromosome 9 ...
... align reference LD on chromosome 9 across populations ...
... 1758 valid SNPs across populations ...
... MCMC ...
--- iter-100 ---
--- iter-200 ---
--- iter-300 ---
--- iter-400 ---
--- iter-500 ---
--- iter-600 ---
--- iter-700 ---
--- iter-800 ---
--- iter-900 ---
--- iter-1000 ---
... Estimated global shrinkage parameter: 8.00e-02 ...
... Done ...
##### process chromosome 10 #####
... parse reference file: PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... 69126 SNPs on chromosome 10 read from PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... parse bim file: SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... 8824 SNPs on chromosome 10 read from SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1969 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1969 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR reference LD on chromosome 10 ...
... parse EUR reference LD on chromosome 10 ...
... align reference LD on chromosome 10 across populations ...
... 1969 valid SNPs across populations ...
... MCMC ...
--- iter-100 ---
--- iter-200 ---
--- iter-300 ---
--- iter-400 ---
--- iter-500 ---
--- iter-600 ---
--- iter-700 ---
--- iter-800 ---
--- iter-900 ---
--- iter-1000 ---
... Estimated global shrinkage parameter: 7.03e-02 ...
... Done ...
##### process chromosome 11 #####
... parse reference file: PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... 66399 SNPs on chromosome 11 read from PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... parse bim file: SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... 8420 SNPs on chromosome 11 read from SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1725 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1725 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR reference LD on chromosome 11 ...
... parse EUR reference LD on chromosome 11 ...
... align reference LD on chromosome 11 across populations ...
... 1725 valid SNPs across populations ...
... MCMC ...
--- iter-100 ---
--- iter-200 ---
--- iter-300 ---
--- iter-400 ---
--- iter-500 ---
--- iter-600 ---
--- iter-700 ---
--- iter-800 ---
--- iter-900 ---
--- iter-1000 ---
... Estimated global shrinkage parameter: 8.12e-02 ...
... Done ...
##### process chromosome 12 #####
... parse reference file: PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... 64628 SNPs on chromosome 12 read from PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... parse bim file: SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... 8198 SNPs on chromosome 12 read from SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1866 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1866 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR reference LD on chromosome 12 ...
... parse EUR reference LD on chromosome 12 ...
... align reference LD on chromosome 12 across populations ...
... 1866 valid SNPs across populations ...
... MCMC ...
--- iter-100 ---
--- iter-200 ---
--- iter-300 ---
--- iter-400 ---
--- iter-500 ---
--- iter-600 ---
--- iter-700 ---
--- iter-800 ---
--- iter-900 ---
--- iter-1000 ---
... Estimated global shrinkage parameter: 8.35e-02 ...
... Done ...
##### process chromosome 13 #####
... parse reference file: PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... 48845 SNPs on chromosome 13 read from PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... parse bim file: SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... 6350 SNPs on chromosome 13 read from SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1389 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1389 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR reference LD on chromosome 13 ...
... parse EUR reference LD on chromosome 13 ...
... align reference LD on chromosome 13 across populations ...
... 1389 valid SNPs across populations ...
... MCMC ...
--- iter-100 ---
--- iter-200 ---
--- iter-300 ---
--- iter-400 ---
--- iter-500 ---
--- iter-600 ---
--- iter-700 ---
--- iter-800 ---
--- iter-900 ---
--- iter-1000 ---
... Estimated global shrinkage parameter: 6.90e-02 ...
... Done ...
##### process chromosome 14 #####
... parse reference file: PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... 42847 SNPs on chromosome 14 read from PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... parse bim file: SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... 5742 SNPs on chromosome 14 read from SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1262 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1262 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR reference LD on chromosome 14 ...
... parse EUR reference LD on chromosome 14 ...
... align reference LD on chromosome 14 across populations ...
... 1262 valid SNPs across populations ...
... MCMC ...
--- iter-100 ---
--- iter-200 ---
--- iter-300 ---
--- iter-400 ---
--- iter-500 ---
--- iter-600 ---
--- iter-700 ---
--- iter-800 ---
--- iter-900 ---
--- iter-1000 ---
... Estimated global shrinkage parameter: 7.60e-02 ...
... Done ...
##### process chromosome 15 #####
... parse reference file: PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... 39395 SNPs on chromosome 15 read from PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... parse bim file: SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... 5569 SNPs on chromosome 15 read from SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1222 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1222 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR reference LD on chromosome 15 ...
... parse EUR reference LD on chromosome 15 ...
... align reference LD on chromosome 15 across populations ...
... 1222 valid SNPs across populations ...
... MCMC ...
--- iter-100 ---
--- iter-200 ---
--- iter-300 ---
--- iter-400 ---
--- iter-500 ---
--- iter-600 ---
--- iter-700 ---
--- iter-800 ---
--- iter-900 ---
--- iter-1000 ---
... Estimated global shrinkage parameter: 8.26e-02 ...
... Done ...
##### process chromosome 16 #####
... parse reference file: PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... 40563 SNPs on chromosome 16 read from PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... parse bim file: SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... 6069 SNPs on chromosome 16 read from SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1283 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1283 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR reference LD on chromosome 16 ...
... parse EUR reference LD on chromosome 16 ...
... align reference LD on chromosome 16 across populations ...
... 1283 valid SNPs across populations ...
... MCMC ...
--- iter-100 ---
--- iter-200 ---
--- iter-300 ---
--- iter-400 ---
--- iter-500 ---
--- iter-600 ---
--- iter-700 ---
--- iter-800 ---
--- iter-900 ---
--- iter-1000 ---
... Estimated global shrinkage parameter: 7.18e-02 ...
... Done ...
##### process chromosome 17 #####
... parse reference file: PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... 36009 SNPs on chromosome 17 read from PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... parse bim file: SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... 5723 SNPs on chromosome 17 read from SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1306 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1306 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR reference LD on chromosome 17 ...
... parse EUR reference LD on chromosome 17 ...
... align reference LD on chromosome 17 across populations ...
... 1306 valid SNPs across populations ...
... MCMC ...
--- iter-100 ---
--- iter-200 ---
--- iter-300 ---
--- iter-400 ---
--- iter-500 ---
--- iter-600 ---
--- iter-700 ---
--- iter-800 ---
--- iter-900 ---
--- iter-1000 ---
... Estimated global shrinkage parameter: 7.79e-02 ...
... Done ...
##### process chromosome 18 #####
... parse reference file: PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... 38591 SNPs on chromosome 18 read from PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... parse bim file: SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... 5578 SNPs on chromosome 18 read from SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1191 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1191 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR reference LD on chromosome 18 ...
... parse EUR reference LD on chromosome 18 ...
... align reference LD on chromosome 18 across populations ...
... 1191 valid SNPs across populations ...
... MCMC ...
--- iter-100 ---
--- iter-200 ---
--- iter-300 ---
--- iter-400 ---
--- iter-500 ---
--- iter-600 ---
--- iter-700 ---
--- iter-800 ---
--- iter-900 ---
--- iter-1000 ---
... Estimated global shrinkage parameter: 7.28e-02 ...
... Done ...
##### process chromosome 19 #####
... parse reference file: PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... 24713 SNPs on chromosome 19 read from PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... parse bim file: SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... 4364 SNPs on chromosome 19 read from SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1108 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1108 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR reference LD on chromosome 19 ...
... parse EUR reference LD on chromosome 19 ...
... align reference LD on chromosome 19 across populations ...
... 1108 valid SNPs across populations ...
... MCMC ...
--- iter-100 ---
--- iter-200 ---
--- iter-300 ---
--- iter-400 ---
--- iter-500 ---
--- iter-600 ---
--- iter-700 ---
--- iter-800 ---
--- iter-900 ---
--- iter-1000 ---
... Estimated global shrinkage parameter: 8.05e-02 ...
... Done ...
##### process chromosome 20 #####
... parse reference file: PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... 33863 SNPs on chromosome 20 read from PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... parse bim file: SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... 4916 SNPs on chromosome 20 read from SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1135 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 1135 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR reference LD on chromosome 20 ...
... parse EUR reference LD on chromosome 20 ...
... align reference LD on chromosome 20 across populations ...
... 1135 valid SNPs across populations ...
... MCMC ...
--- iter-100 ---
--- iter-200 ---
--- iter-300 ---
--- iter-400 ---
--- iter-500 ---
--- iter-600 ---
--- iter-700 ---
--- iter-800 ---
--- iter-900 ---
--- iter-1000 ---
... Estimated global shrinkage parameter: 7.53e-02 ...
... Done ...
##### process chromosome 21 #####
... parse reference file: PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... 18073 SNPs on chromosome 21 read from PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... parse bim file: SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... 2811 SNPs on chromosome 21 read from SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 612 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 612 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR reference LD on chromosome 21 ...
... parse EUR reference LD on chromosome 21 ...
... align reference LD on chromosome 21 across populations ...
... 612 valid SNPs across populations ...
... MCMC ...
--- iter-100 ---
--- iter-200 ---
--- iter-300 ---
--- iter-400 ---
--- iter-500 ---
--- iter-600 ---
--- iter-700 ---
--- iter-800 ---
--- iter-900 ---
--- iter-1000 ---
... Estimated global shrinkage parameter: 6.40e-02 ...
... Done ...
##### process chromosome 22 #####
... parse reference file: PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... 18944 SNPs on chromosome 22 read from PRScsx_reference_panel/ldblk_1kg_eur/snpinfo_mult_1kg_hm3 ...
... parse bim file: SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... 2831 SNPs on chromosome 22 read from SampleData1/Fold_0/train_data.QC.clumped.pruned.bim ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 743 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR sumstats file: SampleData1/SampleData1.PRSCSx ...
... 499617 SNPs read from SampleData1/SampleData1.PRSCSx ...
... 743 common SNPs in the EUR reference, EUR sumstats, and validation set ...
... parse EUR reference LD on chromosome 22 ...
... parse EUR reference LD on chromosome 22 ...
... align reference LD on chromosome 22 across populations ...
... 743 valid SNPs across populations ...
... MCMC ...
--- iter-100 ---
--- iter-200 ---
--- iter-300 ---
--- iter-400 ---
--- iter-500 ---
--- iter-600 ---
--- iter-700 ---
--- iter-800 ---
--- iter-900 ---
--- iter-1000 ---
... Estimated global shrinkage parameter: 9.46e-02 ...
... Done ...
---------------------------------------------------------------------------
RuntimeError Traceback (most recent call last)
Cell In[14], line 221
219 for burning_iteration in burning_iterations:
220 for thining_iteration in thining_iterations:
--> 221 transform_prscs_data(folddirec, newtrainfilename, p,ref_dir,phi,va,vb,number_of_iteration,burning_iteration,thining_iteration, str(p1_val), str(p2_val), str(p3_val), str(c1_val), str(c2_val), str(c3_val), "PRScsx", pvaluefile)
Cell In[14], line 113, in transform_prscs_data(traindirec, newtrainfilename, numberofpca, ref_dir, phi, va, vb, number_of_iteration, burning_iteration, thining_iteration, p1_val, p2_val, p3_val, c1_val, c2_val, c3_val, Name, pvaluefile)
111 file_list = glob.glob(traindirec+os.sep+"PRSCSx*.txt")
112 sorted_file_list = sorted(file_list, key=lambda x: int(''.join(filter(str.isdigit, x))))
--> 113 raise
115 merged_df = pd.DataFrame()
117 # Iterate through the files
RuntimeError: No active exception to reraise
Repeat the process for each fold.#
Change the foldnumber variable.
#foldnumber = sys.argv[1]
foldnumber = "0" # Setting 'foldnumber' to "0"
Or uncomment the following line:
# foldnumber = sys.argv[1]
python PRScsx.py 0
python PRScsx.py 1
python PRScsx.py 2
python PRScsx.py 3
python PRScsx.py 4
The following files should exist after the execution:
SampleData1/Fold_0/PRScsx/Results.csvSampleData1/Fold_1/PRScsx/Results.csvSampleData1/Fold_2/PRScsx/Results.csvSampleData1/Fold_3/PRScsx/Results.csvSampleData1/Fold_4/PRScsx/Results.csv
Check the results file for each fold.#
import os
result_directory = "PRScsx"
# List of file names to check for existence
f = [
"./"+filedirec+"/Fold_0"+os.sep+result_directory+"Results.csv",
"./"+filedirec+"/Fold_1"+os.sep+result_directory+"Results.csv",
"./"+filedirec+"/Fold_2"+os.sep+result_directory+"Results.csv",
"./"+filedirec+"/Fold_3"+os.sep+result_directory+"Results.csv",
"./"+filedirec+"/Fold_4"+os.sep+result_directory+"Results.csv",
]
# Loop through each file name in the list
for loop in range(0,5):
# Check if the file exists in the specified directory for the given fold
if os.path.exists(filedirec+os.sep+"Fold_"+str(loop)+os.sep+result_directory+os.sep+"Results.csv"):
temp = pd.read_csv(filedirec+os.sep+"Fold_"+str(loop)+os.sep+result_directory+os.sep+"Results.csv")
print("Fold_",loop, "Yes, the file exists.")
#print(temp.head())
print("Number of P-values processed: ",len(temp))
# Print a message indicating that the file exists
else:
# Print a message indicating that the file does not exist
print("Fold_",loop, "No, the file does not exist.")
Fold_ 0 Yes, the file exists.
Number of P-values processed: 80
Fold_ 1 Yes, the file exists.
Number of P-values processed: 80
Fold_ 2 Yes, the file exists.
Number of P-values processed: 80
Fold_ 3 Yes, the file exists.
Number of P-values processed: 80
Fold_ 4 Yes, the file exists.
Number of P-values processed: 80
Sum the results for each fold.#
print("We have to ensure when we sum the entries across all Folds, the same rows are merged!")
def sum_and_average_columns(data_frames):
"""Sum and average numerical columns across multiple DataFrames, and keep non-numerical columns unchanged."""
# Initialize DataFrame to store the summed results for numerical columns
summed_df = pd.DataFrame()
non_numerical_df = pd.DataFrame()
for df in data_frames:
# Identify numerical and non-numerical columns
numerical_cols = df.select_dtypes(include=[np.number]).columns
non_numerical_cols = df.select_dtypes(exclude=[np.number]).columns
# Sum numerical columns
if summed_df.empty:
summed_df = pd.DataFrame(0, index=range(len(df)), columns=numerical_cols)
summed_df[numerical_cols] = summed_df[numerical_cols].add(df[numerical_cols], fill_value=0)
# Keep non-numerical columns (take the first non-numerical entry for each column)
if non_numerical_df.empty:
non_numerical_df = df[non_numerical_cols]
else:
non_numerical_df[non_numerical_cols] = non_numerical_df[non_numerical_cols].combine_first(df[non_numerical_cols])
# Divide the summed values by the number of dataframes to get the average
averaged_df = summed_df / len(data_frames)
# Combine numerical and non-numerical DataFrames
result_df = pd.concat([averaged_df, non_numerical_df], axis=1)
return result_df
from functools import reduce
import os
import pandas as pd
from functools import reduce
def find_common_rows(allfoldsframe):
# Define the performance columns that need to be excluded
performance_columns = [
'Train_null_model', 'Train_pure_prs', 'Train_best_model',
'Test_pure_prs', 'Test_null_model', 'Test_best_model'
]
important_columns = [
'clump_p1',
'clump_r2',
'clump_kb',
'p_window_size',
'p_slide_size',
'p_LD_threshold',
'pvalue',
'referencepanel',
'PRSice-2_Model',
'effectsizes',
'h2model',
"PRScs_ref_dir" ,
"PRScs_phi" ,
"PRScs_va" ,
"PRScs_vb" ,
"PRScs_number_of_iteration" ,
"PRScs_burning_iteration" ,
"PRScs_thining_iteration" ,
'numberofpca',
'tempalpha',
'l1weight',
]
# Function to remove performance columns from a DataFrame
def drop_performance_columns(df):
return df.drop(columns=performance_columns, errors='ignore')
def get_important_columns(df ):
existing_columns = [col for col in important_columns if col in df.columns]
if existing_columns:
return df[existing_columns].copy()
else:
return pd.DataFrame()
# Drop performance columns from all DataFrames in the list
allfoldsframe_dropped = [drop_performance_columns(df) for df in allfoldsframe]
# Get the important columns.
allfoldsframe_dropped = [get_important_columns(df) for df in allfoldsframe_dropped]
# Iteratively find common rows and track unique and common rows
common_rows = allfoldsframe_dropped[0]
for i in range(1, len(allfoldsframe_dropped)):
# Get the next DataFrame
next_df = allfoldsframe_dropped[i]
# Count unique rows in the current DataFrame and the next DataFrame
unique_in_common = common_rows.shape[0]
unique_in_next = next_df.shape[0]
# Find common rows between the current common_rows and the next DataFrame
common_rows = pd.merge(common_rows, next_df, how='inner')
# Count the common rows after merging
common_count = common_rows.shape[0]
# Print the unique and common row counts
print(f"Iteration {i}:")
print(f"Unique rows in current common DataFrame: {unique_in_common}")
print(f"Unique rows in next DataFrame: {unique_in_next}")
print(f"Common rows after merge: {common_count}\n")
# Now that we have the common rows, extract these from the original DataFrames
extracted_common_rows_frames = []
for original_df in allfoldsframe:
# Merge the common rows with the original DataFrame, keeping only the rows that match the common rows
extracted_common_rows = pd.merge(common_rows, original_df, how='inner', on=common_rows.columns.tolist())
# Add the DataFrame with the extracted common rows to the list
extracted_common_rows_frames.append(extracted_common_rows)
# Print the number of rows in the common DataFrames
for i, df in enumerate(extracted_common_rows_frames):
print(f"DataFrame {i + 1} with extracted common rows has {df.shape[0]} rows.")
# Return the list of DataFrames with extracted common rows
return extracted_common_rows_frames
# Example usage (assuming allfoldsframe is populated as shown earlier):
allfoldsframe = []
# Loop through each file name in the list
for loop in range(0, 5):
# Check if the file exists in the specified directory for the given fold
file_path = os.path.join(filedirec, "Fold_" + str(loop), result_directory, "Results.csv")
if os.path.exists(file_path):
allfoldsframe.append(pd.read_csv(file_path))
# Print a message indicating that the file exists
print("Fold_", loop, "Yes, the file exists.")
else:
# Print a message indicating that the file does not exist
print("Fold_", loop, "No, the file does not exist.")
# Find the common rows across all folds and return the list of extracted common rows
extracted_common_rows_list = find_common_rows(allfoldsframe)
# Sum the values column-wise
# For string values, do not sum it the values are going to be the same for each fold.
# Only sum the numeric values.
divided_result = sum_and_average_columns(extracted_common_rows_list)
print(divided_result)
We have to ensure when we sum the entries across all Folds, the same rows are merged!
Fold_ 0 Yes, the file exists.
Fold_ 1 Yes, the file exists.
Fold_ 2 Yes, the file exists.
Fold_ 3 Yes, the file exists.
Fold_ 4 Yes, the file exists.
Iteration 1:
Unique rows in current common DataFrame: 80
Unique rows in next DataFrame: 80
Common rows after merge: 80
Iteration 2:
Unique rows in current common DataFrame: 80
Unique rows in next DataFrame: 80
Common rows after merge: 80
Iteration 3:
Unique rows in current common DataFrame: 80
Unique rows in next DataFrame: 80
Common rows after merge: 80
Iteration 4:
Unique rows in current common DataFrame: 80
Unique rows in next DataFrame: 80
Common rows after merge: 80
DataFrame 1 with extracted common rows has 80 rows.
DataFrame 2 with extracted common rows has 80 rows.
DataFrame 3 with extracted common rows has 80 rows.
DataFrame 4 with extracted common rows has 80 rows.
DataFrame 5 with extracted common rows has 80 rows.
clump_p1 clump_r2 clump_kb p_window_size p_slide_size p_LD_threshold \
0 1.0 0.1 200.0 200.0 50.0 0.25
1 1.0 0.1 200.0 200.0 50.0 0.25
2 1.0 0.1 200.0 200.0 50.0 0.25
3 1.0 0.1 200.0 200.0 50.0 0.25
4 1.0 0.1 200.0 200.0 50.0 0.25
.. ... ... ... ... ... ...
75 1.0 0.1 200.0 200.0 50.0 0.25
76 1.0 0.1 200.0 200.0 50.0 0.25
77 1.0 0.1 200.0 200.0 50.0 0.25
78 1.0 0.1 200.0 200.0 50.0 0.25
79 1.0 0.1 200.0 200.0 50.0 0.25
pvalue PRScs_va PRScs_vb PRScs_number_of_iteration ... \
0 1.000000e-10 1.0 0.5 1000.0 ...
1 3.359818e-10 1.0 0.5 1000.0 ...
2 1.128838e-09 1.0 0.5 1000.0 ...
3 3.792690e-09 1.0 0.5 1000.0 ...
4 1.274275e-08 1.0 0.5 1000.0 ...
.. ... ... ... ... ...
75 7.847600e-03 1.0 0.5 1000.0 ...
76 2.636651e-02 1.0 0.5 1000.0 ...
77 8.858668e-02 1.0 0.5 1000.0 ...
78 2.976351e-01 1.0 0.5 1000.0 ...
79 1.000000e+00 1.0 0.5 1000.0 ...
tempalpha l1weight Train_pure_prs Train_null_model Train_best_model \
0 0.1 0.1 0.000055 0.23001 0.237074
1 0.1 0.1 0.000049 0.23001 0.237388
2 0.1 0.1 0.000049 0.23001 0.240353
3 0.1 0.1 0.000049 0.23001 0.243013
4 0.1 0.1 0.000047 0.23001 0.245642
.. ... ... ... ... ...
75 0.1 0.1 0.000007 0.23001 0.271152
76 0.1 0.1 0.000005 0.23001 0.271224
77 0.1 0.1 0.000003 0.23001 0.271225
78 0.1 0.1 0.000002 0.23001 0.271280
79 0.1 0.1 0.000001 0.23001 0.271260
Test_pure_prs Test_null_model Test_best_model PRScs_ref_dir PRScs_phi
0 0.000034 0.118692 0.114407 ldblk_1kg_eur auto
1 0.000028 0.118692 0.115888 ldblk_1kg_eur auto
2 0.000034 0.118692 0.117896 ldblk_1kg_eur auto
3 0.000040 0.118692 0.117961 ldblk_1kg_eur auto
4 0.000038 0.118692 0.118853 ldblk_1kg_eur auto
.. ... ... ... ... ...
75 0.000006 0.118692 0.159427 ldblk_ukbb_eur 1e-6
76 0.000004 0.118692 0.159905 ldblk_ukbb_eur 1e-6
77 0.000003 0.118692 0.159836 ldblk_ukbb_eur 1e-6
78 0.000002 0.118692 0.159832 ldblk_ukbb_eur 1e-6
79 0.000001 0.118692 0.159857 ldblk_ukbb_eur 1e-6
[80 rows x 23 columns]
/tmp/ipykernel_2352549/1121511899.py:24: SettingWithCopyWarning:
A value is trying to be set on a copy of a slice from a DataFrame.
Try using .loc[row_indexer,col_indexer] = value instead
See the caveats in the documentation: https://pandas.pydata.org/pandas-docs/stable/user_guide/indexing.html#returning-a-view-versus-a-copy
non_numerical_df[non_numerical_cols] = non_numerical_df[non_numerical_cols].combine_first(df[non_numerical_cols])
/tmp/ipykernel_2352549/1121511899.py:24: SettingWithCopyWarning:
A value is trying to be set on a copy of a slice from a DataFrame.
Try using .loc[row_indexer,col_indexer] = value instead
See the caveats in the documentation: https://pandas.pydata.org/pandas-docs/stable/user_guide/indexing.html#returning-a-view-versus-a-copy
non_numerical_df[non_numerical_cols] = non_numerical_df[non_numerical_cols].combine_first(df[non_numerical_cols])
/tmp/ipykernel_2352549/1121511899.py:24: SettingWithCopyWarning:
A value is trying to be set on a copy of a slice from a DataFrame.
Try using .loc[row_indexer,col_indexer] = value instead
See the caveats in the documentation: https://pandas.pydata.org/pandas-docs/stable/user_guide/indexing.html#returning-a-view-versus-a-copy
non_numerical_df[non_numerical_cols] = non_numerical_df[non_numerical_cols].combine_first(df[non_numerical_cols])
/tmp/ipykernel_2352549/1121511899.py:24: SettingWithCopyWarning:
A value is trying to be set on a copy of a slice from a DataFrame.
Try using .loc[row_indexer,col_indexer] = value instead
See the caveats in the documentation: https://pandas.pydata.org/pandas-docs/stable/user_guide/indexing.html#returning-a-view-versus-a-copy
non_numerical_df[non_numerical_cols] = non_numerical_df[non_numerical_cols].combine_first(df[non_numerical_cols])
Results#
1. Reporting Based on Best Training Performance:#
One can report the results based on the best performance of the training data. For example, if for a specific combination of hyperparameters, the training performance is high, report the corresponding test performance.
Example code:
df = divided_result.sort_values(by='Train_best_model', ascending=False) print(df.iloc[0].to_markdown())
Binary Phenotypes Result Analysis#
You can find the performance quality for binary phenotype using the following template:
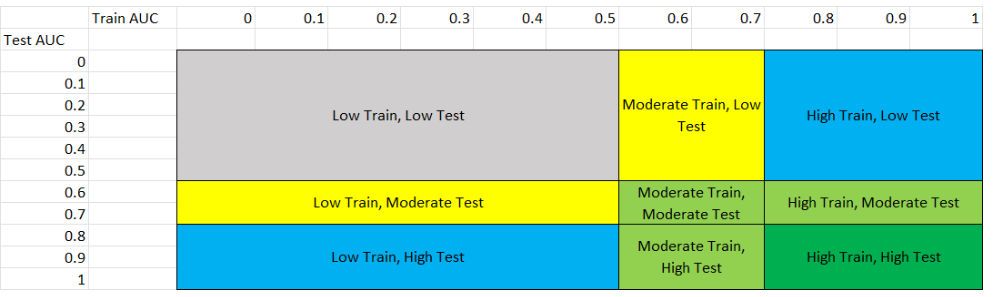
This figure shows the 8 different scenarios that can exist in the results, and the following table explains each scenario.
We classified performance based on the following table:
Performance Level |
Range |
|---|---|
Low Performance |
0 to 0.5 |
Moderate Performance |
0.6 to 0.7 |
High Performance |
0.8 to 1 |
You can match the performance based on the following scenarios:
Scenario |
What’s Happening |
Implication |
|---|---|---|
High Test, High Train |
The model performs well on both training and test datasets, effectively learning the underlying patterns. |
The model is well-tuned, generalizes well, and makes accurate predictions on both datasets. |
High Test, Moderate Train |
The model generalizes well but may not be fully optimized on training data, missing some underlying patterns. |
The model is fairly robust but may benefit from further tuning or more training to improve its learning. |
High Test, Low Train |
An unusual scenario, potentially indicating data leakage or overestimation of test performance. |
The model’s performance is likely unreliable; investigate potential data issues or random noise. |
Moderate Test, High Train |
The model fits the training data well but doesn’t generalize as effectively, capturing only some test patterns. |
The model is slightly overfitting; adjustments may be needed to improve generalization on unseen data. |
Moderate Test, Moderate Train |
The model shows balanced but moderate performance on both datasets, capturing some patterns but missing others. |
The model is moderately fitting; further improvements could be made in both training and generalization. |
Moderate Test, Low Train |
The model underperforms on training data and doesn’t generalize well, leading to moderate test performance. |
The model may need more complexity, additional features, or better training to improve on both datasets. |
Low Test, High Train |
The model overfits the training data, performing poorly on the test set. |
The model doesn’t generalize well; simplifying the model or using regularization may help reduce overfitting. |
Low Test, Low Train |
The model performs poorly on both training and test datasets, failing to learn the data patterns effectively. |
The model is underfitting; it may need more complexity, additional features, or more data to improve performance. |
Recommendations for Publishing Results#
When publishing results, scenarios with moderate train and moderate test performance can be used for complex phenotypes or diseases. However, results showing high train and moderate test, high train and high test, and moderate train and high test are recommended.
For most phenotypes, results typically fall in the moderate train and moderate test performance category.
Continuous Phenotypes Result Analysis#
You can find the performance quality for continuous phenotypes using the following template:
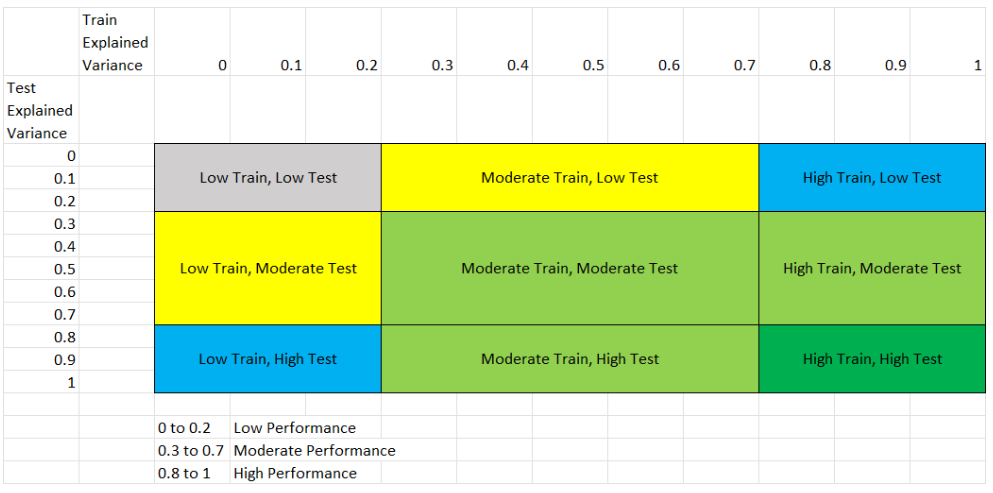
This figure shows the 8 different scenarios that can exist in the results, and the following table explains each scenario.
We classified performance based on the following table:
Performance Level |
Range |
|---|---|
Low Performance |
0 to 0.2 |
Moderate Performance |
0.3 to 0.7 |
High Performance |
0.8 to 1 |
You can match the performance based on the following scenarios:
Scenario |
What’s Happening |
Implication |
|---|---|---|
High Test, High Train |
The model performs well on both training and test datasets, effectively learning the underlying patterns. |
The model is well-tuned, generalizes well, and makes accurate predictions on both datasets. |
High Test, Moderate Train |
The model generalizes well but may not be fully optimized on training data, missing some underlying patterns. |
The model is fairly robust but may benefit from further tuning or more training to improve its learning. |
High Test, Low Train |
An unusual scenario, potentially indicating data leakage or overestimation of test performance. |
The model’s performance is likely unreliable; investigate potential data issues or random noise. |
Moderate Test, High Train |
The model fits the training data well but doesn’t generalize as effectively, capturing only some test patterns. |
The model is slightly overfitting; adjustments may be needed to improve generalization on unseen data. |
Moderate Test, Moderate Train |
The model shows balanced but moderate performance on both datasets, capturing some patterns but missing others. |
The model is moderately fitting; further improvements could be made in both training and generalization. |
Moderate Test, Low Train |
The model underperforms on training data and doesn’t generalize well, leading to moderate test performance. |
The model may need more complexity, additional features, or better training to improve on both datasets. |
Low Test, High Train |
The model overfits the training data, performing poorly on the test set. |
The model doesn’t generalize well; simplifying the model or using regularization may help reduce overfitting. |
Low Test, Low Train |
The model performs poorly on both training and test datasets, failing to learn the data patterns effectively. |
The model is underfitting; it may need more complexity, additional features, or more data to improve performance. |
Recommendations for Publishing Results#
When publishing results, scenarios with moderate train and moderate test performance can be used for complex phenotypes or diseases. However, results showing high train and moderate test, high train and high test, and moderate train and high test are recommended.
For most continuous phenotypes, results typically fall in the moderate train and moderate test performance category.
2. Reporting Generalized Performance:#
One can also report the generalized performance by calculating the difference between the training and test performance, and the sum of the test and training performance. Report the result or hyperparameter combination for which the sum is high and the difference is minimal.
Example code:
df = divided_result.copy() df['Difference'] = abs(df['Train_best_model'] - df['Test_best_model']) df['Sum'] = df['Train_best_model'] + df['Test_best_model'] sorted_df = df.sort_values(by=['Sum', 'Difference'], ascending=[False, True]) print(sorted_df.iloc[0].to_markdown())
3. Reporting Hyperparameters Affecting Test and Train Performance:#
Find the hyperparameters that have more than one unique value and calculate their correlation with the following columns to understand how they are affecting the performance of train and test sets:
Train_null_modelTrain_pure_prsTrain_best_modelTest_pure_prsTest_null_modelTest_best_model
4. Other Analysis#
Once you have the results, you can find how hyperparameters affect the model performance.
Analysis, like overfitting and underfitting, can be performed as well.
The way you are going to report the results can vary.
Results can be visualized, and other patterns in the data can be explored.
import pandas as pd
import seaborn as sns
import matplotlib.pyplot as plt
%matplotlib notebook
import matplotlib
import numpy as np
import matplotlib.pyplot as plt
df = divided_result.sort_values(by='Train_best_model', ascending=False)
print("1. Reporting Based on Best Training Performance:\n")
print(df.iloc[0].to_markdown())
df = divided_result.copy()
# Plot Train and Test best models against p-values
plt.figure(figsize=(10, 6))
plt.plot(df['pvalue'], df['Train_best_model'], label='Train_best_model', marker='o', color='royalblue')
plt.plot(df['pvalue'], df['Test_best_model'], label='Test_best_model', marker='o', color='darkorange')
# Highlight the p-value where both train and test are high
best_index = df[['Train_best_model']].sum(axis=1).idxmax()
best_pvalue = df.loc[best_index, 'pvalue']
best_train = df.loc[best_index, 'Train_best_model']
best_test = df.loc[best_index, 'Test_best_model']
# Use dark colors for the circles
plt.scatter(best_pvalue, best_train, color='darkred', s=100, label=f'Best Performance (Train)', edgecolor='black', zorder=5)
plt.scatter(best_pvalue, best_test, color='darkblue', s=100, label=f'Best Performance (Test)', edgecolor='black', zorder=5)
# Annotate the best performance with p-value, train, and test values
plt.text(best_pvalue, best_train, f'p={best_pvalue:.4g}\nTrain={best_train:.4g}', ha='right', va='bottom', fontsize=9, color='darkred')
plt.text(best_pvalue, best_test, f'p={best_pvalue:.4g}\nTest={best_test:.4g}', ha='right', va='top', fontsize=9, color='darkblue')
# Calculate Difference and Sum
df['Difference'] = abs(df['Train_best_model'] - df['Test_best_model'])
df['Sum'] = df['Train_best_model'] + df['Test_best_model']
# Sort the DataFrame
sorted_df = df.sort_values(by=['Sum', 'Difference'], ascending=[False, True])
#sorted_df = df.sort_values(by=[ 'Difference','Sum'], ascending=[ True,False])
# Highlight the general performance
general_index = sorted_df.index[0]
general_pvalue = sorted_df.loc[general_index, 'pvalue']
general_train = sorted_df.loc[general_index, 'Train_best_model']
general_test = sorted_df.loc[general_index, 'Test_best_model']
plt.scatter(general_pvalue, general_train, color='darkgreen', s=150, label='General Performance (Train)', edgecolor='black', zorder=6)
plt.scatter(general_pvalue, general_test, color='darkorange', s=150, label='General Performance (Test)', edgecolor='black', zorder=6)
# Annotate the general performance with p-value, train, and test values
plt.text(general_pvalue, general_train, f'p={general_pvalue:.4g}\nTrain={general_train:.4g}', ha='left', va='bottom', fontsize=9, color='darkgreen')
plt.text(general_pvalue, general_test, f'p={general_pvalue:.4g}\nTest={general_test:.4g}', ha='left', va='top', fontsize=9, color='darkorange')
# Add labels and legend
plt.xlabel('p-value')
plt.ylabel('Model Performance')
plt.title('Train vs Test Best Models')
plt.legend()
plt.show()
print("2. Reporting Generalized Performance:\n")
df = divided_result.copy()
df['Difference'] = abs(df['Train_best_model'] - df['Test_best_model'])
df['Sum'] = df['Train_best_model'] + df['Test_best_model']
sorted_df = df.sort_values(by=['Sum', 'Difference'], ascending=[False, True])
print(sorted_df.iloc[0].to_markdown())
print("3. Reporting the correlation of hyperparameters and the performance of 'Train_null_model', 'Train_pure_prs', 'Train_best_model', 'Test_pure_prs', 'Test_null_model', and 'Test_best_model':\n")
print("3. For string hyperparameters, we used one-hot encoding to find the correlation between string hyperparameters and 'Train_null_model', 'Train_pure_prs', 'Train_best_model', 'Test_pure_prs', 'Test_null_model', and 'Test_best_model'.")
print("3. We performed this analysis for those hyperparameters that have more than one unique value.")
correlation_columns = [
'Train_null_model', 'Train_pure_prs', 'Train_best_model',
'Test_pure_prs', 'Test_null_model', 'Test_best_model'
]
hyperparams = [col for col in divided_result.columns if len(divided_result[col].unique()) > 1]
hyperparams = list(set(hyperparams+correlation_columns))
# Separate numeric and string columns
numeric_hyperparams = [col for col in hyperparams if pd.api.types.is_numeric_dtype(divided_result[col])]
string_hyperparams = [col for col in hyperparams if pd.api.types.is_string_dtype(divided_result[col])]
# Encode string columns using one-hot encoding
divided_result_encoded = pd.get_dummies(divided_result, columns=string_hyperparams)
# Combine numeric hyperparams with the new one-hot encoded columns
encoded_columns = [col for col in divided_result_encoded.columns if col.startswith(tuple(string_hyperparams))]
hyperparams = numeric_hyperparams + encoded_columns
# Calculate correlations
correlations = divided_result_encoded[hyperparams].corr()
# Display correlation of hyperparameters with train/test performance columns
hyperparam_correlations = correlations.loc[hyperparams, correlation_columns]
hyperparam_correlations = hyperparam_correlations.fillna(0)
# Plotting the correlation heatmap
plt.figure(figsize=(12, 8))
ax = sns.heatmap(hyperparam_correlations, annot=True, cmap='viridis', fmt='.2f', cbar=True)
ax.set_xticklabels(ax.get_xticklabels(), rotation=90, ha='right')
# Rotate y-axis labels to horizontal
#ax.set_yticklabels(ax.get_yticklabels(), rotation=0, va='center')
plt.title('Correlation of Hyperparameters with Train/Test Performance')
plt.show()
sns.set_theme(style="whitegrid") # Choose your preferred style
pairplot = sns.pairplot(divided_result_encoded[hyperparams],hue = 'Test_best_model', palette='viridis')
# Adjust the figure size
pairplot.fig.set_size_inches(15, 15) # You can adjust the size as needed
for ax in pairplot.axes.flatten():
ax.set_xlabel(ax.get_xlabel(), rotation=90, ha='right') # X-axis labels vertical
#ax.set_ylabel(ax.get_ylabel(), rotation=0, va='bottom') # Y-axis labels horizontal
# Show the plot
plt.show()
1. Reporting Based on Best Training Performance:
| | 58 |
|:--------------------------|:----------------------|
| clump_p1 | 1.0 |
| clump_r2 | 0.1 |
| clump_kb | 200.0 |
| p_window_size | 200.0 |
| p_slide_size | 50.0 |
| p_LD_threshold | 0.25 |
| pvalue | 0.2976351441631313 |
| PRScs_va | 1.0 |
| PRScs_vb | 0.5 |
| PRScs_number_of_iteration | 1000.0 |
| PRScs_burning_iteration | 500.0 |
| PRScs_thining_iteration | 5.0 |
| numberofpca | 6.0 |
| tempalpha | 0.1 |
| l1weight | 0.1 |
| Train_pure_prs | 4.264393649089371e-06 |
| Train_null_model | 0.23001030414198947 |
| Train_best_model | 0.32766124537953606 |
| Test_pure_prs | 4.316253223857202e-06 |
| Test_null_model | 0.11869244971793831 |
| Test_best_model | 0.23582861703918362 |
| PRScs_ref_dir | ldblk_ukbb_eur |
| PRScs_phi | auto |

2. Reporting Generalized Performance:
| | 58 |
|:--------------------------|:----------------------|
| clump_p1 | 1.0 |
| clump_r2 | 0.1 |
| clump_kb | 200.0 |
| p_window_size | 200.0 |
| p_slide_size | 50.0 |
| p_LD_threshold | 0.25 |
| pvalue | 0.2976351441631313 |
| PRScs_va | 1.0 |
| PRScs_vb | 0.5 |
| PRScs_number_of_iteration | 1000.0 |
| PRScs_burning_iteration | 500.0 |
| PRScs_thining_iteration | 5.0 |
| numberofpca | 6.0 |
| tempalpha | 0.1 |
| l1weight | 0.1 |
| Train_pure_prs | 4.264393649089371e-06 |
| Train_null_model | 0.23001030414198947 |
| Train_best_model | 0.32766124537953606 |
| Test_pure_prs | 4.316253223857202e-06 |
| Test_null_model | 0.11869244971793831 |
| Test_best_model | 0.23582861703918362 |
| PRScs_ref_dir | ldblk_ukbb_eur |
| PRScs_phi | auto |
| Difference | 0.09183262834035244 |
| Sum | 0.5634898624187197 |
3. Reporting the correlation of hyperparameters and the performance of 'Train_null_model', 'Train_pure_prs', 'Train_best_model', 'Test_pure_prs', 'Test_null_model', and 'Test_best_model':
3. For string hyperparameters, we used one-hot encoding to find the correlation between string hyperparameters and 'Train_null_model', 'Train_pure_prs', 'Train_best_model', 'Test_pure_prs', 'Test_null_model', and 'Test_best_model'.
3. We performed this analysis for those hyperparameters that have more than one unique value.

